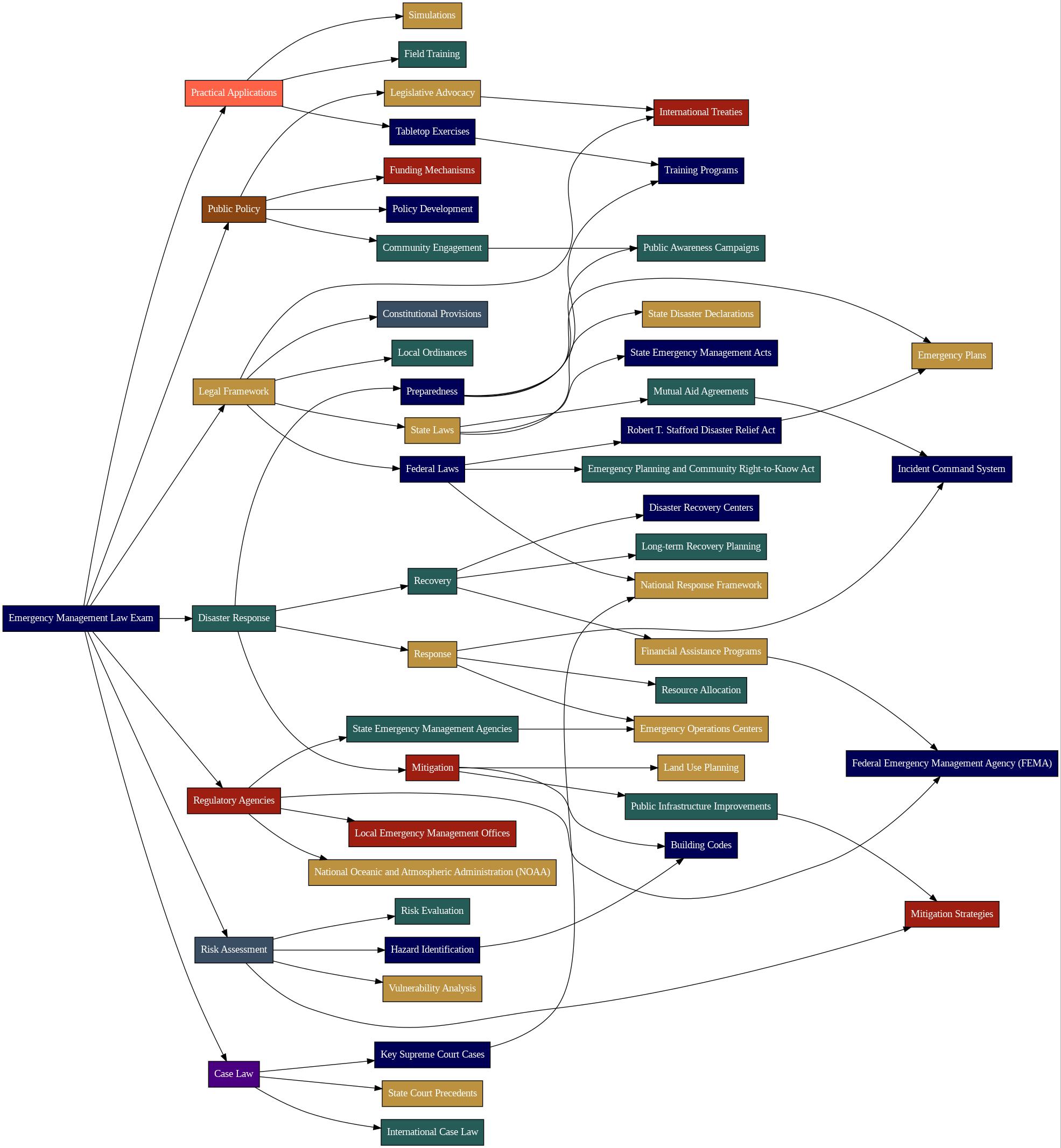
Quiz-summary
0 of 30 questions completed
Questions:
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
Information
Premium Practice Questions
You have already completed the quiz before. Hence you can not start it again.
Quiz is loading...
You must sign in or sign up to start the quiz.
You have to finish following quiz, to start this quiz:
Results
0 of 30 questions answered correctly
Your time:
Time has elapsed
Categories
- Not categorized 0%
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- Answered
- Review
-
Question 1 of 30
1. Question
A biotechnology firm develops a novel formulation containing a proprietary blend of prebiotics and probiotics, designed to enhance gut microbiome balance. The company intends to market this product to consumers seeking to improve their digestive well-being. They plan to include statements on the packaging such as “Supports a healthy digestive tract” and “Promotes optimal gut function.” Considering the regulatory frameworks governing food and drug law, under which classification would the company most likely be able to legally market its product with these specific claims, assuming adequate substantiation for the claims exists?
Correct
The core of this question lies in understanding the regulatory distinctions between a dietary supplement and a conventional food product, particularly concerning the claims made. A product marketed as a “dietary supplement” under the Dietary Supplement Health and Education Act of 1994 (DSHEA) can make structure/function claims, provided they are substantiated and accompanied by a disclaimer. Structure/function claims describe the role of a nutrient or dietary ingredient intended to affect the normal structure or function of the human body (e.g., “calcium builds strong bones”). Conversely, a product classified as a conventional food, regulated under the Federal Food, Drug, and Cosmetic Act (FD&C Act), is generally intended for ingestion as food. While conventional foods can bear nutritional claims and certain health claims (which require pre-market approval by the FDA), they cannot make structure/function claims in the same manner as dietary supplements. The scenario describes a product containing probiotics and prebiotics, intended for “gut health support” and “improved digestion.” These phrases directly relate to the function of the gastrointestinal system. If the product is marketed as a dietary supplement, such claims are permissible with proper substantiation and disclaimer. If it were marketed as a conventional food, these claims would be problematic, potentially leading to misbranding, as conventional foods are not typically permitted to make such direct functional assertions without specific FDA authorization (e.g., a qualified health claim, which has a different standard than structure/function claims). The key differentiator is the intended use and the regulatory pathway chosen by the manufacturer. The product’s formulation (probiotics and prebiotics) is common in both categories, but the marketing claims and intended classification are paramount. Therefore, the ability to legally market a product with these specific claims hinges on its classification as a dietary supplement, allowing for structure/function claims.
Incorrect
The core of this question lies in understanding the regulatory distinctions between a dietary supplement and a conventional food product, particularly concerning the claims made. A product marketed as a “dietary supplement” under the Dietary Supplement Health and Education Act of 1994 (DSHEA) can make structure/function claims, provided they are substantiated and accompanied by a disclaimer. Structure/function claims describe the role of a nutrient or dietary ingredient intended to affect the normal structure or function of the human body (e.g., “calcium builds strong bones”). Conversely, a product classified as a conventional food, regulated under the Federal Food, Drug, and Cosmetic Act (FD&C Act), is generally intended for ingestion as food. While conventional foods can bear nutritional claims and certain health claims (which require pre-market approval by the FDA), they cannot make structure/function claims in the same manner as dietary supplements. The scenario describes a product containing probiotics and prebiotics, intended for “gut health support” and “improved digestion.” These phrases directly relate to the function of the gastrointestinal system. If the product is marketed as a dietary supplement, such claims are permissible with proper substantiation and disclaimer. If it were marketed as a conventional food, these claims would be problematic, potentially leading to misbranding, as conventional foods are not typically permitted to make such direct functional assertions without specific FDA authorization (e.g., a qualified health claim, which has a different standard than structure/function claims). The key differentiator is the intended use and the regulatory pathway chosen by the manufacturer. The product’s formulation (probiotics and prebiotics) is common in both categories, but the marketing claims and intended classification are paramount. Therefore, the ability to legally market a product with these specific claims hinges on its classification as a dietary supplement, allowing for structure/function claims.
-
Question 2 of 30
2. Question
A company is marketing a new product containing a blend of botanical extracts and vitamins. The product’s packaging prominently features claims such as “Proven to lower cholesterol levels by 15% in clinical studies” and “Helps prevent the onset of Type 2 Diabetes.” The company asserts that the product is a dietary supplement intended to support overall wellness. Under the Federal Food, Drug, and Cosmetic Act (FDCA), what is the most accurate regulatory classification for this product, and what is the primary implication for its marketing?
Correct
The core of this question lies in understanding the regulatory distinction between a dietary supplement and a drug under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to diagnose, cure, mitigate, treat, or prevent disease is classified as a drug. The labeling of a product with claims that it can treat a specific medical condition, such as reducing the risk of a particular disease or affecting the structure or function of the body in relation to a disease, firmly places it within the drug definition. Specifically, claims that a substance can “reduce the risk of cardiovascular disease” or “treat hypertension” are characteristic drug claims. Dietary supplements, while permitted to make structure/function claims (e.g., “supports cardiovascular health” or “helps maintain healthy blood pressure”), cannot make these disease-oriented claims. The FDCA, particularly Section 201(g)(1)(B), defines a drug as an article intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals. Therefore, a product making such disease-specific claims, regardless of its ingredients or intended use for general well-being, would be regulated as a drug. The presence of a New Drug Application (NDA) or an Abbreviated New Drug Application (ANDA) is the pathway for legitimate drug marketing. Without such approval, marketing a product with drug claims is a violation of the FDCA. The scenario describes a product with ingredients commonly found in dietary supplements but with labeling that crosses the line into drug territory by asserting disease treatment and risk reduction. This necessitates regulation as a drug, requiring an approved marketing authorization.
Incorrect
The core of this question lies in understanding the regulatory distinction between a dietary supplement and a drug under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to diagnose, cure, mitigate, treat, or prevent disease is classified as a drug. The labeling of a product with claims that it can treat a specific medical condition, such as reducing the risk of a particular disease or affecting the structure or function of the body in relation to a disease, firmly places it within the drug definition. Specifically, claims that a substance can “reduce the risk of cardiovascular disease” or “treat hypertension” are characteristic drug claims. Dietary supplements, while permitted to make structure/function claims (e.g., “supports cardiovascular health” or “helps maintain healthy blood pressure”), cannot make these disease-oriented claims. The FDCA, particularly Section 201(g)(1)(B), defines a drug as an article intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals. Therefore, a product making such disease-specific claims, regardless of its ingredients or intended use for general well-being, would be regulated as a drug. The presence of a New Drug Application (NDA) or an Abbreviated New Drug Application (ANDA) is the pathway for legitimate drug marketing. Without such approval, marketing a product with drug claims is a violation of the FDCA. The scenario describes a product with ingredients commonly found in dietary supplements but with labeling that crosses the line into drug territory by asserting disease treatment and risk reduction. This necessitates regulation as a drug, requiring an approved marketing authorization.
-
Question 3 of 30
3. Question
A biotechnology firm, “AlgaNova,” has developed a novel food ingredient derived from genetically modified algae, which they claim significantly boosts antioxidant levels in consumer products. AlgaNova has conducted extensive in-vitro and animal studies demonstrating the ingredient’s purported health benefits and safety profile. They intend to market this ingredient for widespread use in beverages and dietary supplements without seeking prior FDA approval or submitting a GRAS notification. Which of the following regulatory actions by the FDA would be most appropriate given AlgaNova’s intended market entry strategy?
Correct
The scenario describes a situation where a novel food ingredient, derived from genetically modified algae, is being introduced into the market. The manufacturer claims it possesses enhanced antioxidant properties and has undergone extensive laboratory testing to support these claims. However, the ingredient has not been subject to a formal pre-market approval process by the Food and Drug Administration (FDA) for novel food ingredients, nor has it been evaluated under the Generally Recognized as Safe (GRAS) notification process. The core issue revolves around the regulatory pathway for such a product under the Federal Food, Drug, and Cosmetic Act (FDCA). Under the FDCA, specifically Section 409 concerning food additives, any substance that is intentionally added to food and does not have a history of safe use prior to January 1, 1958, or is not GRAS, is considered a food additive and requires pre-market approval through a Food Additive Petition (FAP). The genetically modified nature of the algae, and the claim of enhanced properties, suggests it likely falls outside the scope of existing GRAS affirmations for common food ingredients. While the manufacturer conducted laboratory testing, this does not substitute for the rigorous scientific review and public comment period mandated for an FAP. The absence of an FAP or a GRAS notification means the ingredient has not received FDA authorization for its intended use. Therefore, its marketing and sale would constitute a violation of the FDCA, rendering the food adulterated or misbranded. The correct regulatory approach would have been to either file an FAP or obtain a GRAS notification, demonstrating the safety of the ingredient for its intended use.
Incorrect
The scenario describes a situation where a novel food ingredient, derived from genetically modified algae, is being introduced into the market. The manufacturer claims it possesses enhanced antioxidant properties and has undergone extensive laboratory testing to support these claims. However, the ingredient has not been subject to a formal pre-market approval process by the Food and Drug Administration (FDA) for novel food ingredients, nor has it been evaluated under the Generally Recognized as Safe (GRAS) notification process. The core issue revolves around the regulatory pathway for such a product under the Federal Food, Drug, and Cosmetic Act (FDCA). Under the FDCA, specifically Section 409 concerning food additives, any substance that is intentionally added to food and does not have a history of safe use prior to January 1, 1958, or is not GRAS, is considered a food additive and requires pre-market approval through a Food Additive Petition (FAP). The genetically modified nature of the algae, and the claim of enhanced properties, suggests it likely falls outside the scope of existing GRAS affirmations for common food ingredients. While the manufacturer conducted laboratory testing, this does not substitute for the rigorous scientific review and public comment period mandated for an FAP. The absence of an FAP or a GRAS notification means the ingredient has not received FDA authorization for its intended use. Therefore, its marketing and sale would constitute a violation of the FDCA, rendering the food adulterated or misbranded. The correct regulatory approach would have been to either file an FAP or obtain a GRAS notification, demonstrating the safety of the ingredient for its intended use.
-
Question 4 of 30
4. Question
Consider a pharmaceutical company that has successfully developed a novel gene-editing therapy designed to correct a specific genetic mutation responsible for a rare, debilitating autoimmune disease. This therapy involves modifying a patient’s own cells ex vivo using advanced CRISPR-Cas9 technology before reinfusion. The company intends to seek FDA approval for this groundbreaking treatment. Which of the following regulatory pathways represents the most appropriate initial submission for this product, given its biological nature and therapeutic intent?
Correct
The scenario describes a situation where a novel therapeutic agent, developed through advanced gene-editing techniques, is being considered for market approval. This agent targets a rare genetic disorder with no existing treatments. The core regulatory challenge lies in assessing the safety and efficacy of a product derived from cutting-edge biotechnology, particularly concerning potential off-target genetic modifications and long-term effects. The Food and Drug Administration (FDA) employs a tiered approach to evaluating such innovations. For biologics, the primary pathway involves a Biologics License Application (BLA). However, given the unique nature of gene-editing technology, the FDA might also leverage its authority under the Federal Food, Drug, and Cosmetic Act (FDCA) to ensure comprehensive safety evaluation. The BLA process, as outlined in the Public Health Service Act (PHSA) and further detailed in FDA regulations (e.g., 21 CFR Part 600), requires extensive preclinical data, including in vitro and in vivo studies demonstrating the product’s mechanism of action, purity, potency, and stability. Crucially, clinical trials must be conducted in phases (Phase 1 for safety, Phase 2 for efficacy and dose-ranging, Phase 3 for confirmation of efficacy and monitoring of adverse events) to gather robust evidence. For a gene-editing therapy, particular emphasis would be placed on demonstrating the precision of the editing, the absence of unintended genomic alterations, and the durability of the therapeutic effect. The regulatory framework also mandates post-marketing surveillance, including pharmacovigilance and adverse event reporting, to monitor the product’s performance in the broader patient population. The question probes the most appropriate initial regulatory submission pathway for a product that, while derived from a biological source and intended for therapeutic use, utilizes a novel manufacturing process (gene editing). While a New Drug Application (NDA) is for chemically synthesized drugs, and a medical device pathway is for devices, a product that modifies human cells for therapeutic purposes falls under the purview of biologics. Therefore, the initial submission would be a BLA, as it is designed for products derived from living organisms or their components, which includes gene therapies. The complexity of the technology might necessitate additional data requirements or specific review processes beyond the standard BLA, but the fundamental classification points to a BLA.
Incorrect
The scenario describes a situation where a novel therapeutic agent, developed through advanced gene-editing techniques, is being considered for market approval. This agent targets a rare genetic disorder with no existing treatments. The core regulatory challenge lies in assessing the safety and efficacy of a product derived from cutting-edge biotechnology, particularly concerning potential off-target genetic modifications and long-term effects. The Food and Drug Administration (FDA) employs a tiered approach to evaluating such innovations. For biologics, the primary pathway involves a Biologics License Application (BLA). However, given the unique nature of gene-editing technology, the FDA might also leverage its authority under the Federal Food, Drug, and Cosmetic Act (FDCA) to ensure comprehensive safety evaluation. The BLA process, as outlined in the Public Health Service Act (PHSA) and further detailed in FDA regulations (e.g., 21 CFR Part 600), requires extensive preclinical data, including in vitro and in vivo studies demonstrating the product’s mechanism of action, purity, potency, and stability. Crucially, clinical trials must be conducted in phases (Phase 1 for safety, Phase 2 for efficacy and dose-ranging, Phase 3 for confirmation of efficacy and monitoring of adverse events) to gather robust evidence. For a gene-editing therapy, particular emphasis would be placed on demonstrating the precision of the editing, the absence of unintended genomic alterations, and the durability of the therapeutic effect. The regulatory framework also mandates post-marketing surveillance, including pharmacovigilance and adverse event reporting, to monitor the product’s performance in the broader patient population. The question probes the most appropriate initial regulatory submission pathway for a product that, while derived from a biological source and intended for therapeutic use, utilizes a novel manufacturing process (gene editing). While a New Drug Application (NDA) is for chemically synthesized drugs, and a medical device pathway is for devices, a product that modifies human cells for therapeutic purposes falls under the purview of biologics. Therefore, the initial submission would be a BLA, as it is designed for products derived from living organisms or their components, which includes gene therapies. The complexity of the technology might necessitate additional data requirements or specific review processes beyond the standard BLA, but the fundamental classification points to a BLA.
-
Question 5 of 30
5. Question
A biotechnology firm has developed a novel therapeutic agent consisting of genetically engineered bacteriophages intended to combat a specific strain of multidrug-resistant bacteria. This agent is manufactured using complex cell culture and purification processes. Which regulatory pathway would the U.S. Food and Drug Administration (FDA) primarily require the firm to follow for marketing approval of this innovative biological product?
Correct
The scenario describes a novel therapeutic agent, a genetically modified bacteriophage designed to target a specific antibiotic-resistant bacterial strain. The development of such an agent falls under the purview of biologics regulation due to its biological origin and complex manufacturing process. The primary pathway for approval of a new biologic product in the United States is through a Biologics License Application (BLA), as mandated by the Public Health Service Act (PHSA) and further detailed by FDA regulations. The BLA process requires extensive preclinical and clinical data demonstrating the safety and efficacy of the product. While the Food, Drug, and Cosmetic Act (FD&C Act) governs drugs, biologics are distinct and are regulated under the PHSA, which is administered by the Center for Biologics Evaluation and Research (CBER) within the FDA. Investigational New Drug (IND) applications are required for clinical trials, but the ultimate marketing approval for a biologic is the BLA. The Orphan Drug Act provides incentives for developing treatments for rare diseases, which might apply if the target bacterial strain is rare, but it does not alter the fundamental regulatory pathway. The 510(k) process is for medical devices, and New Drug Applications (NDAs) are for chemically synthesized drugs, neither of which applies to this genetically engineered biological product. Therefore, the correct regulatory pathway for marketing approval is the BLA.
Incorrect
The scenario describes a novel therapeutic agent, a genetically modified bacteriophage designed to target a specific antibiotic-resistant bacterial strain. The development of such an agent falls under the purview of biologics regulation due to its biological origin and complex manufacturing process. The primary pathway for approval of a new biologic product in the United States is through a Biologics License Application (BLA), as mandated by the Public Health Service Act (PHSA) and further detailed by FDA regulations. The BLA process requires extensive preclinical and clinical data demonstrating the safety and efficacy of the product. While the Food, Drug, and Cosmetic Act (FD&C Act) governs drugs, biologics are distinct and are regulated under the PHSA, which is administered by the Center for Biologics Evaluation and Research (CBER) within the FDA. Investigational New Drug (IND) applications are required for clinical trials, but the ultimate marketing approval for a biologic is the BLA. The Orphan Drug Act provides incentives for developing treatments for rare diseases, which might apply if the target bacterial strain is rare, but it does not alter the fundamental regulatory pathway. The 510(k) process is for medical devices, and New Drug Applications (NDAs) are for chemically synthesized drugs, neither of which applies to this genetically engineered biological product. Therefore, the correct regulatory pathway for marketing approval is the BLA.
-
Question 6 of 30
6. Question
A company develops a new topical product named “Glow-Up Serum,” advertised with claims that it “visibly reduces the appearance of fine lines and wrinkles by stimulating collagen production” and “enhances skin elasticity for a firmer, more youthful look.” The product contains a novel peptide complex. Considering the regulatory definitions under the Federal Food, Drug, and Cosmetic Act (FDCA), how would the Food and Drug Administration (FDA) most likely classify “Glow-Up Serum” based on these marketing claims?
Correct
The core of this question lies in understanding the regulatory distinction between a “drug” and a “cosmetic” under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to affect the structure or function of the body, or to treat or prevent disease, is classified as a drug. Conversely, a cosmetic is intended for cleansing, beautifying, promoting attractiveness, or altering the appearance without affecting the body’s structure or function. The product in question, “Glow-Up Serum,” is marketed with claims of “reducing the appearance of fine lines and wrinkles by stimulating collagen production” and “improving skin elasticity.” These claims directly relate to altering the skin’s structure (collagen production) and function (elasticity), thereby classifying it as a drug. Specifically, the claim of stimulating collagen production falls under the purview of affecting the body’s structure. Therefore, Glow-Up Serum would be regulated as a drug, requiring an approved New Drug Application (NDA) or an Investigational New Drug (IND) application if it were a new drug being tested. The other options represent incorrect classifications. A dietary supplement is regulated under a different framework, primarily by the Dietary Supplement Health and Education Act of 1994 (DSHEA), and its claims are limited to structure/function claims that do not diagnose, treat, cure, or prevent disease. A medical device has a distinct definition and regulatory pathway under the FDCA, typically involving premarket notification (510(k)) or premarket approval (PMA). A general cosmetic, while subject to labeling and safety requirements, cannot make claims that alter the structure or function of the body. The regulatory framework for drugs is significantly more stringent due to the potential impact on public health.
Incorrect
The core of this question lies in understanding the regulatory distinction between a “drug” and a “cosmetic” under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to affect the structure or function of the body, or to treat or prevent disease, is classified as a drug. Conversely, a cosmetic is intended for cleansing, beautifying, promoting attractiveness, or altering the appearance without affecting the body’s structure or function. The product in question, “Glow-Up Serum,” is marketed with claims of “reducing the appearance of fine lines and wrinkles by stimulating collagen production” and “improving skin elasticity.” These claims directly relate to altering the skin’s structure (collagen production) and function (elasticity), thereby classifying it as a drug. Specifically, the claim of stimulating collagen production falls under the purview of affecting the body’s structure. Therefore, Glow-Up Serum would be regulated as a drug, requiring an approved New Drug Application (NDA) or an Investigational New Drug (IND) application if it were a new drug being tested. The other options represent incorrect classifications. A dietary supplement is regulated under a different framework, primarily by the Dietary Supplement Health and Education Act of 1994 (DSHEA), and its claims are limited to structure/function claims that do not diagnose, treat, cure, or prevent disease. A medical device has a distinct definition and regulatory pathway under the FDCA, typically involving premarket notification (510(k)) or premarket approval (PMA). A general cosmetic, while subject to labeling and safety requirements, cannot make claims that alter the structure or function of the body. The regulatory framework for drugs is significantly more stringent due to the potential impact on public health.
-
Question 7 of 30
7. Question
A biotechnology firm develops a topical serum marketed as “AuraGlow.” The product’s labeling prominently features claims such as “visibly diminishes the appearance of age-related skin laxity by promoting cellular regeneration” and “enhances dermal firmness through targeted peptide delivery.” The firm asserts that these effects are achieved by the serum’s active ingredients interacting with skin cells to boost natural repair mechanisms. Based on the Federal Food, Drug, and Cosmetic Act (FDCA), how would the U.S. Food and Drug Administration (FDA) most likely classify AuraGlow given these marketing claims?
Correct
The core of this question lies in understanding the regulatory distinction between a “drug” and a “cosmetic” under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to affect the structure or function of the body, or to treat or prevent disease, is classified as a drug. Conversely, a product intended for cleansing, beautifying, promoting attractiveness, or altering the appearance, without affecting the body’s structure or function, is considered a cosmetic. In this scenario, the “Revitalizing Serum” is marketed with claims of “reducing the appearance of fine lines and wrinkles by stimulating collagen production” and “improving skin elasticity.” The claim of “stimulating collagen production” directly implicates a physiological effect on the skin’s structure, aiming to alter its natural processes. Similarly, “improving skin elasticity” suggests a change in the physical properties of the skin, which falls under the purview of drug regulation rather than mere cosmetic alteration. Therefore, the product’s claims push it beyond the definition of a cosmetic and into the realm of a drug, requiring an approved New Drug Application (NDA) before marketing. The other options present plausible but incorrect classifications. A dietary supplement is regulated under the Dietary Supplement Health and Education Act (DSHEA) and has different labeling and marketing requirements, typically focusing on nutrient content and structure/function claims that do not treat or prevent disease. A medical device is defined by its intended use, which is typically to diagnose, cure, mitigate, treat, or prevent disease, or to affect the structure or function of the body, but it does not achieve its primary intended purposes through chemical action within or on the body, nor is it metabolized. While some topical products can be considered medical devices, the claims here are more indicative of a drug’s pharmacological action. A food additive is a substance intended for use in food, and its regulation focuses on safety and functionality within the food matrix, which is clearly not the case here.
Incorrect
The core of this question lies in understanding the regulatory distinction between a “drug” and a “cosmetic” under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to affect the structure or function of the body, or to treat or prevent disease, is classified as a drug. Conversely, a product intended for cleansing, beautifying, promoting attractiveness, or altering the appearance, without affecting the body’s structure or function, is considered a cosmetic. In this scenario, the “Revitalizing Serum” is marketed with claims of “reducing the appearance of fine lines and wrinkles by stimulating collagen production” and “improving skin elasticity.” The claim of “stimulating collagen production” directly implicates a physiological effect on the skin’s structure, aiming to alter its natural processes. Similarly, “improving skin elasticity” suggests a change in the physical properties of the skin, which falls under the purview of drug regulation rather than mere cosmetic alteration. Therefore, the product’s claims push it beyond the definition of a cosmetic and into the realm of a drug, requiring an approved New Drug Application (NDA) before marketing. The other options present plausible but incorrect classifications. A dietary supplement is regulated under the Dietary Supplement Health and Education Act (DSHEA) and has different labeling and marketing requirements, typically focusing on nutrient content and structure/function claims that do not treat or prevent disease. A medical device is defined by its intended use, which is typically to diagnose, cure, mitigate, treat, or prevent disease, or to affect the structure or function of the body, but it does not achieve its primary intended purposes through chemical action within or on the body, nor is it metabolized. While some topical products can be considered medical devices, the claims here are more indicative of a drug’s pharmacological action. A food additive is a substance intended for use in food, and its regulation focuses on safety and functionality within the food matrix, which is clearly not the case here.
-
Question 8 of 30
8. Question
Consider a novel sweetener, “Sweet-All,” developed by the “Nutri-Innovate” company. Nutri-Innovate has compiled extensive scientific data, including toxicological studies and metabolic pathway analyses, demonstrating Sweet-All’s safety for human consumption at proposed usage levels. This data was submitted to the FDA for review, and the agency has issued an affirmation of GRAS status for Sweet-All under specific conditions of use. If Nutri-Innovate wishes to market Sweet-All as an ingredient in baked goods, which of the following regulatory actions is *not* required for compliance with the Federal Food, Drug, and Cosmetic Act?
Correct
The core of this question lies in understanding the regulatory distinctions between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance is considered a food additive if it is not GRAS and is intended for use in food. The FDCA defines a food additive as “any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food.” Crucially, if a substance has a history of safe use prior to January 1, 1958, or has been demonstrated to be safe through scientific procedures, it can be considered GRAS. The key differentiator is the regulatory pathway for approval and the basis for its safety determination. Food additives require premarket approval through a Food Additive Petition (FAP), which involves extensive scientific data demonstrating safety under intended conditions of use. GRAS status, conversely, can be achieved through scientific procedures or, for substances used before 1958, through common use in food. The distinction is not merely about the presence of a substance in food, but the legal basis for its safety and intended use. Therefore, a substance that has undergone rigorous scientific review and has been affirmed as GRAS by the FDA, or is considered GRAS based on common use prior to 1958, does not require a Food Additive Petition. The regulatory framework prioritizes safety, and GRAS status represents a recognized pathway to market for substances deemed safe without the need for a formal FAP.
Incorrect
The core of this question lies in understanding the regulatory distinctions between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance is considered a food additive if it is not GRAS and is intended for use in food. The FDCA defines a food additive as “any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food.” Crucially, if a substance has a history of safe use prior to January 1, 1958, or has been demonstrated to be safe through scientific procedures, it can be considered GRAS. The key differentiator is the regulatory pathway for approval and the basis for its safety determination. Food additives require premarket approval through a Food Additive Petition (FAP), which involves extensive scientific data demonstrating safety under intended conditions of use. GRAS status, conversely, can be achieved through scientific procedures or, for substances used before 1958, through common use in food. The distinction is not merely about the presence of a substance in food, but the legal basis for its safety and intended use. Therefore, a substance that has undergone rigorous scientific review and has been affirmed as GRAS by the FDA, or is considered GRAS based on common use prior to 1958, does not require a Food Additive Petition. The regulatory framework prioritizes safety, and GRAS status represents a recognized pathway to market for substances deemed safe without the need for a formal FAP.
-
Question 9 of 30
9. Question
A biotechnology firm, “BioGlow Innovations,” develops a novel topical serum. The product’s marketing materials highlight its ability to “visibly reduce the appearance of fine lines and wrinkles” and “promote a radiant complexion,” typical cosmetic claims. However, the serum also contains a proprietary peptide complex that the company’s research suggests stimulates fibroblast activity and collagen synthesis, thereby “aiding in the repair of sun-damaged skin and mitigating the effects of photoaging.” The firm intends to distribute this product through beauty supply stores and online retailers, without seeking prior FDA approval. Under the Federal Food, Drug, and Cosmetic Act, what is the most likely regulatory classification of BioGlow Innovations’ serum, and what is the primary basis for this determination?
Correct
The core issue revolves around the regulatory classification of a product intended for both cosmetic and therapeutic purposes. The Federal Food, Drug, and Cosmetic Act (FD&C Act) defines a “drug” as an article intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in humans or other animals, or an article (other than food) intended to affect the structure or any function of the body. A “cosmetic” is defined as an article intended to be rubbed, poured, sprinkled, or sprayed on, introduced into, or otherwise applied to the human body or any part thereof for cleansing, beautifying, promoting attractiveness, or altering the appearance. When a product possesses characteristics of both categories, its intended use, as determined by labeling, advertising, and other evidence, dictates its regulatory classification. If the product’s claims or marketing suggest it treats a medical condition or affects bodily function beyond mere beautification, it will be regulated as a drug. The presence of an active ingredient that is not generally recognized as safe and effective for its intended cosmetic use, and which is also intended to affect the structure or function of the body, further solidifies its drug classification. Therefore, a product marketed with claims of both skin rejuvenation and the mitigation of a specific dermatological condition, containing an ingredient not established as safe and effective for cosmetic purposes but known to influence cellular regeneration, would fall under the stringent drug regulatory framework, requiring an approved New Drug Application (NDA) or an Investigational New Drug (IND) application for clinical testing.
Incorrect
The core issue revolves around the regulatory classification of a product intended for both cosmetic and therapeutic purposes. The Federal Food, Drug, and Cosmetic Act (FD&C Act) defines a “drug” as an article intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in humans or other animals, or an article (other than food) intended to affect the structure or any function of the body. A “cosmetic” is defined as an article intended to be rubbed, poured, sprinkled, or sprayed on, introduced into, or otherwise applied to the human body or any part thereof for cleansing, beautifying, promoting attractiveness, or altering the appearance. When a product possesses characteristics of both categories, its intended use, as determined by labeling, advertising, and other evidence, dictates its regulatory classification. If the product’s claims or marketing suggest it treats a medical condition or affects bodily function beyond mere beautification, it will be regulated as a drug. The presence of an active ingredient that is not generally recognized as safe and effective for its intended cosmetic use, and which is also intended to affect the structure or function of the body, further solidifies its drug classification. Therefore, a product marketed with claims of both skin rejuvenation and the mitigation of a specific dermatological condition, containing an ingredient not established as safe and effective for cosmetic purposes but known to influence cellular regeneration, would fall under the stringent drug regulatory framework, requiring an approved New Drug Application (NDA) or an Investigational New Drug (IND) application for clinical testing.
-
Question 10 of 30
10. Question
A biotechnology firm, “NutriGen Innovations,” has developed a novel, synthesized compound named “Xylosorb.” This compound is intended to be incorporated into a variety of baked goods to improve moisture retention and extend shelf life. Preliminary internal studies suggest Xylosorb is safe for consumption at the proposed levels. However, Xylosorb has not been previously approved by the Food and Drug Administration (FDA) for any food use, nor has it achieved Generally Recognized as Safe (GRAS) status for its intended application as a humectant in baked goods. To legally market baked goods containing Xylosorb, what regulatory pathway must NutriGen Innovations primarily pursue?
Correct
The core of this question lies in understanding the regulatory distinction between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance that is intended for use in food and has not been previously sanctioned as a food additive or is not GRAS is subject to the food additive petition process. The FDCA defines a food additive as “any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food.” However, it explicitly excludes “a GRAS substance.” Therefore, if a novel compound, “Xylosorb,” is intended to function as a humectant in baked goods and lacks prior FDA approval or widespread recognition of safety for this specific use, it would necessitate a food additive petition. The process involves submitting scientific data demonstrating safety for the intended use, which the FDA then reviews. If approved, it is added to the list of permitted food additives. The absence of a GRAS affirmation or prior approval for Xylosorb’s intended humectant function in baked goods dictates the need for this formal petitioning process. The other options represent scenarios that do not align with the initial regulatory pathway for a novel, unapproved substance intended for food use. A post-market surveillance report is for already approved substances, a New Drug Application (NDA) is for pharmaceuticals, and a voluntary recall is a corrective action taken after a product is already on the market, not the initial regulatory hurdle.
Incorrect
The core of this question lies in understanding the regulatory distinction between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance that is intended for use in food and has not been previously sanctioned as a food additive or is not GRAS is subject to the food additive petition process. The FDCA defines a food additive as “any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food.” However, it explicitly excludes “a GRAS substance.” Therefore, if a novel compound, “Xylosorb,” is intended to function as a humectant in baked goods and lacks prior FDA approval or widespread recognition of safety for this specific use, it would necessitate a food additive petition. The process involves submitting scientific data demonstrating safety for the intended use, which the FDA then reviews. If approved, it is added to the list of permitted food additives. The absence of a GRAS affirmation or prior approval for Xylosorb’s intended humectant function in baked goods dictates the need for this formal petitioning process. The other options represent scenarios that do not align with the initial regulatory pathway for a novel, unapproved substance intended for food use. A post-market surveillance report is for already approved substances, a New Drug Application (NDA) is for pharmaceuticals, and a voluntary recall is a corrective action taken after a product is already on the market, not the initial regulatory hurdle.
-
Question 11 of 30
11. Question
Consider a scenario where a biotechnology firm, “AromaGen Corp.,” has developed a novel synthetic compound designed to significantly enhance the umami flavor profile of processed meat products. This compound, designated “UmamiBoost-X,” has undergone preliminary in-vitro testing suggesting a favorable safety profile, but it has not been previously affirmed as Generally Recognized as Safe (GRAS) for consumption in any food category. AromaGen Corp. intends to market UmamiBoost-X for use in a wide array of cured meats, sausages, and meat analogues. Under the Federal Food, Drug, and Cosmetic Act (FDCA), what is the primary regulatory pathway AromaGen Corp. must pursue to legally introduce UmamiBoost-X into the food supply for its intended use?
Correct
The core of this question revolves around the regulatory distinction between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance is considered a food additive if it is not GRAS and is intended for use in food. The FDCA defines a food additive as “any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food.” Crucially, if a substance is intended to affect the characteristics of food, such as preserving it or enhancing its flavor, and it does not meet the criteria for being GRAS, it requires premarket approval through a Food Additive Petition (FAP). The FDCA mandates that a food additive must be approved by the FDA before it can be legally used in food. This approval process involves demonstrating the safety of the substance under its intended conditions of use. Substances that are GRAS are exempt from the food additive provisions of the FDCA. However, the status of a substance as GRAS can be challenged or revoked if new scientific evidence emerges indicating a lack of safety. Therefore, when a substance’s intended use is to impart a specific functional characteristic to food and its GRAS status is uncertain or has been withdrawn, it necessitates a formal regulatory pathway for approval, which is the FAP. The scenario describes a novel flavoring agent intended to enhance the taste profile of baked goods. Without a pre-existing GRAS affirmation for this specific flavoring agent in this application, it falls under the definition of a food additive. Consequently, the appropriate regulatory action is to submit a Food Additive Petition to the FDA for review and approval, ensuring its safety for consumption in the intended food product.
Incorrect
The core of this question revolves around the regulatory distinction between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance is considered a food additive if it is not GRAS and is intended for use in food. The FDCA defines a food additive as “any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food.” Crucially, if a substance is intended to affect the characteristics of food, such as preserving it or enhancing its flavor, and it does not meet the criteria for being GRAS, it requires premarket approval through a Food Additive Petition (FAP). The FDCA mandates that a food additive must be approved by the FDA before it can be legally used in food. This approval process involves demonstrating the safety of the substance under its intended conditions of use. Substances that are GRAS are exempt from the food additive provisions of the FDCA. However, the status of a substance as GRAS can be challenged or revoked if new scientific evidence emerges indicating a lack of safety. Therefore, when a substance’s intended use is to impart a specific functional characteristic to food and its GRAS status is uncertain or has been withdrawn, it necessitates a formal regulatory pathway for approval, which is the FAP. The scenario describes a novel flavoring agent intended to enhance the taste profile of baked goods. Without a pre-existing GRAS affirmation for this specific flavoring agent in this application, it falls under the definition of a food additive. Consequently, the appropriate regulatory action is to submit a Food Additive Petition to the FDA for review and approval, ensuring its safety for consumption in the intended food product.
-
Question 12 of 30
12. Question
A pharmaceutical company has developed “Neuro-Regen,” a promising new drug targeting a rare neurodegenerative disorder that affects an estimated 8,000 individuals in the United States. Following successful completion of Phase II clinical trials that indicated both preliminary efficacy and an acceptable safety profile, the company is exploring regulatory pathways to accelerate its development and eventual market entry. Considering the limited patient population and the significant unmet medical need for this condition, what specific regulatory designation should the company prioritize to leverage incentives for developing treatments for rare diseases?
Correct
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for treating a rare neurodegenerative disorder. The developer has completed Phase II clinical trials demonstrating preliminary efficacy and safety. The disorder affects fewer than 10,000 individuals in the United States. The developer seeks to expedite the approval process due to the unmet medical need and the limited patient population. The correct regulatory pathway for a drug intended to treat a rare disease or condition affecting a small population in the US is Orphan Drug Designation. This designation, granted under the Orphan Drug Act of 1983, provides incentives such as market exclusivity for seven years upon approval, tax credits for qualified clinical research expenses, and assistance with the development process. While the drug is in early stages, the question focuses on the *designation* that facilitates expedited development and market exclusivity for rare diseases. The New Drug Application (NDA) is the formal submission for approval, but the Orphan Drug Designation is a prerequisite for certain incentives and expedited pathways for rare diseases. Fast Track designation is for drugs that treat serious conditions and fill an unmet medical need, but Orphan Drug Designation is specifically tied to the rare disease aspect. Breakthrough Therapy designation is for drugs that demonstrate substantial improvement over available therapies, which may or may not be the case here based solely on Phase II data. Therefore, the most appropriate designation to pursue at this stage, given the rare disease context and desire for expedited development incentives, is Orphan Drug Designation.
Incorrect
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for treating a rare neurodegenerative disorder. The developer has completed Phase II clinical trials demonstrating preliminary efficacy and safety. The disorder affects fewer than 10,000 individuals in the United States. The developer seeks to expedite the approval process due to the unmet medical need and the limited patient population. The correct regulatory pathway for a drug intended to treat a rare disease or condition affecting a small population in the US is Orphan Drug Designation. This designation, granted under the Orphan Drug Act of 1983, provides incentives such as market exclusivity for seven years upon approval, tax credits for qualified clinical research expenses, and assistance with the development process. While the drug is in early stages, the question focuses on the *designation* that facilitates expedited development and market exclusivity for rare diseases. The New Drug Application (NDA) is the formal submission for approval, but the Orphan Drug Designation is a prerequisite for certain incentives and expedited pathways for rare diseases. Fast Track designation is for drugs that treat serious conditions and fill an unmet medical need, but Orphan Drug Designation is specifically tied to the rare disease aspect. Breakthrough Therapy designation is for drugs that demonstrate substantial improvement over available therapies, which may or may not be the case here based solely on Phase II data. Therefore, the most appropriate designation to pursue at this stage, given the rare disease context and desire for expedited development incentives, is Orphan Drug Designation.
-
Question 13 of 30
13. Question
A pharmaceutical company has developed a novel coronary stent coated with a proprietary anticoagulant drug designed to prevent restenosis. This “drug-eluting stent” is intended to deliver the drug directly to the arterial wall over a sustained period. Considering the regulatory frameworks established by the Federal Food, Drug, and Cosmetic Act (FD&C Act) and the Public Health Service Act (PHS Act), which regulatory pathway would the U.S. Food and Drug Administration (FDA) primarily utilize for the approval of this combination product, given that the drug’s pharmacological action is considered the primary mode of action for achieving the intended therapeutic effect?
Correct
The core issue revolves around the regulatory pathway for a novel therapeutic agent that exhibits characteristics of both a drug and a device, specifically a drug-eluting stent. The Federal Food, Drug, and Cosmetic Act (FD&C Act) and the Public Health Service Act (PHS Act) are the primary legislative frameworks governing such products. When a product combines elements regulated under different acts, the FDA employs a “lead constituent element” approach to determine the primary regulatory pathway. In this case, the stent itself is a medical device, but its therapeutic effect is delivered by the drug coating. The FDA’s guidance documents, particularly those addressing combination products, clarify that the regulatory pathway is determined by the constituent element that provides the primary mode of action. For a drug-eluting stent, the drug’s pharmacological action is critical to its therapeutic benefit, making the drug the lead constituent element. Therefore, the product would be regulated as a drug, requiring a New Drug Application (NDA) under the FD&C Act, even though it also incorporates device components. The Investigational New Drug (IND) application process would precede the NDA, allowing for clinical investigation of the drug’s safety and efficacy. While a Premarket Approval (PMA) or Premarket Notification (510(k)) might be relevant for the device component in isolation, the combination product’s primary mode of action dictates the overarching regulatory pathway. The PHS Act primarily governs biologics, which is not the case here. The Food Safety Modernization Act (FSMA) is focused on food safety and is irrelevant to this scenario. The Drug Enforcement Administration (DEA) regulations pertain to controlled substances, which are not mentioned.
Incorrect
The core issue revolves around the regulatory pathway for a novel therapeutic agent that exhibits characteristics of both a drug and a device, specifically a drug-eluting stent. The Federal Food, Drug, and Cosmetic Act (FD&C Act) and the Public Health Service Act (PHS Act) are the primary legislative frameworks governing such products. When a product combines elements regulated under different acts, the FDA employs a “lead constituent element” approach to determine the primary regulatory pathway. In this case, the stent itself is a medical device, but its therapeutic effect is delivered by the drug coating. The FDA’s guidance documents, particularly those addressing combination products, clarify that the regulatory pathway is determined by the constituent element that provides the primary mode of action. For a drug-eluting stent, the drug’s pharmacological action is critical to its therapeutic benefit, making the drug the lead constituent element. Therefore, the product would be regulated as a drug, requiring a New Drug Application (NDA) under the FD&C Act, even though it also incorporates device components. The Investigational New Drug (IND) application process would precede the NDA, allowing for clinical investigation of the drug’s safety and efficacy. While a Premarket Approval (PMA) or Premarket Notification (510(k)) might be relevant for the device component in isolation, the combination product’s primary mode of action dictates the overarching regulatory pathway. The PHS Act primarily governs biologics, which is not the case here. The Food Safety Modernization Act (FSMA) is focused on food safety and is irrelevant to this scenario. The Drug Enforcement Administration (DEA) regulations pertain to controlled substances, which are not mentioned.
-
Question 14 of 30
14. Question
A pharmaceutical company has developed “Neuro-Regen,” a novel agent for a rare neurodegenerative disorder affecting fewer than 200,000 individuals in the U.S. Preclinical studies and Phase 1/2 trials have established a favorable safety profile and demonstrated statistically significant improvement in a key disease biomarker. Phase 3 trials are underway, with preliminary data suggesting a trend towards clinical benefit but not yet achieving statistical significance for the primary endpoint due to a smaller-than-anticipated trial cohort. Which regulatory pathway under the FDCA would most appropriately facilitate the potential for earlier market access for Neuro-Regen, acknowledging the need for post-market confirmation of clinical benefit?
Correct
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for treating a rare neurodegenerative disorder. The manufacturer has conducted extensive preclinical studies demonstrating safety and preliminary efficacy. They have also completed Phase 1 and Phase 2 clinical trials, which provided robust data on safety, tolerability, and a statistically significant improvement in a key biomarker for the disease. However, Phase 3 trials are still ongoing, with preliminary results showing a trend towards clinical benefit but not yet reaching statistical significance for the primary endpoint due to a smaller-than-anticipated patient population in the trial. The disorder affects fewer than 200,000 individuals in the United States. Under the Federal Food, Drug, and Cosmetic Act (FDCA), specifically provisions related to expedited drug approvals, the FDA offers several pathways for drugs intended to treat serious or life-threatening conditions. These pathways are designed to accelerate the availability of promising therapies. Given the rare nature of the disease (orphan drug designation criteria) and the promising, albeit not yet statistically definitive, clinical data, the most appropriate pathway for early consideration of market approval would be the Accelerated Approval pathway. This pathway allows for approval based on surrogate endpoints or intermediate clinical endpoints that are reasonably likely to predict clinical benefit. The ongoing Phase 3 trials would then be required to confirm the clinical benefit post-approval. While Fast Track designation and Breakthrough Therapy designation are also important for accelerating development, they are designations that facilitate interaction and do not, in themselves, grant approval. Priority Review is a designation that shortens the FDA’s review timeline once an application is submitted. Therefore, Accelerated Approval is the regulatory mechanism that best fits the described situation for potential early market access, contingent on post-market confirmatory studies.
Incorrect
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for treating a rare neurodegenerative disorder. The manufacturer has conducted extensive preclinical studies demonstrating safety and preliminary efficacy. They have also completed Phase 1 and Phase 2 clinical trials, which provided robust data on safety, tolerability, and a statistically significant improvement in a key biomarker for the disease. However, Phase 3 trials are still ongoing, with preliminary results showing a trend towards clinical benefit but not yet reaching statistical significance for the primary endpoint due to a smaller-than-anticipated patient population in the trial. The disorder affects fewer than 200,000 individuals in the United States. Under the Federal Food, Drug, and Cosmetic Act (FDCA), specifically provisions related to expedited drug approvals, the FDA offers several pathways for drugs intended to treat serious or life-threatening conditions. These pathways are designed to accelerate the availability of promising therapies. Given the rare nature of the disease (orphan drug designation criteria) and the promising, albeit not yet statistically definitive, clinical data, the most appropriate pathway for early consideration of market approval would be the Accelerated Approval pathway. This pathway allows for approval based on surrogate endpoints or intermediate clinical endpoints that are reasonably likely to predict clinical benefit. The ongoing Phase 3 trials would then be required to confirm the clinical benefit post-approval. While Fast Track designation and Breakthrough Therapy designation are also important for accelerating development, they are designations that facilitate interaction and do not, in themselves, grant approval. Priority Review is a designation that shortens the FDA’s review timeline once an application is submitted. Therefore, Accelerated Approval is the regulatory mechanism that best fits the described situation for potential early market access, contingent on post-market confirmatory studies.
-
Question 15 of 30
15. Question
A biotechnology firm, “CogniVita Labs,” has developed a novel oral capsule containing a proprietary blend of nootropics and a standardized extract of *Ginkgo biloba*. The product is marketed as a “cognitive enhancer” designed to “improve memory recall and concentration.” *Ginkgo biloba* extract is known in traditional medicine for its purported circulatory and cognitive benefits. CogniVita Labs intends to sell this product as a dietary supplement. However, the specific claims made about “improving memory recall and concentration” and the inclusion of an ingredient with established medicinal associations raise questions about its regulatory status under the Federal Food, Drug, and Cosmetic Act (FDCA). Which regulatory pathway is most appropriate for this product, considering the potential overlap between food and drug definitions?
Correct
The core issue revolves around the regulatory classification of a novel product intended for oral consumption that purports to enhance cognitive function and also contains a botanical extract with established medicinal properties. The Federal Food, Drug, and Cosmetic Act (FDCA) defines a “drug” as an article intended to affect the structure or function of the body, and also articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease. A “food” is defined as an article used for food or drink for man or other animals, chewing gum, and articles used for components of any such article. The product in question is marketed as a dietary supplement, which is a category of food under the FDCA. However, the claims made about enhancing cognitive function and the presence of a botanical extract with known medicinal effects push it towards a drug classification. Specifically, if the product is intended to diagnose, cure, mitigate, treat, or prevent disease, or to affect the structure or function of the body in a manner beyond normal nutritional support, it would be considered a drug. The phrase “affect the structure or function of the body” is broad. While dietary supplements can make “structure/function claims” (e.g., “supports healthy joints”), these claims must be substantiated and cannot imply disease treatment or prevention. The presence of a botanical extract with documented medicinal properties, coupled with claims of cognitive enhancement, strongly suggests an intent to affect the body’s function in a way that likely crosses the line from a permissible dietary supplement claim to a drug claim. The regulatory burden for drugs is significantly higher, requiring premarket approval through a New Drug Application (NDA) or Biologics License Application (BLA). Dietary supplements, on the other hand, are regulated differently, with the manufacturer responsible for ensuring safety and proper labeling, and the FDA taking action against adulterated or misbranded products. Given the dual nature of the product’s claims and ingredients, the most appropriate regulatory pathway, and the one that aligns with the FDCA’s definitions, is to treat it as a drug. This necessitates an NDA process. The other options are incorrect because: classifying it solely as a dietary supplement ignores the drug-like claims and ingredients; treating it as a cosmetic is inappropriate as it is intended for ingestion and affects internal bodily functions, not external appearance; and the Public Health Service Act primarily governs biologics and certain public health activities, not the classification of food-like products with drug claims. Therefore, the product’s characteristics necessitate regulation as a drug, requiring an NDA.
Incorrect
The core issue revolves around the regulatory classification of a novel product intended for oral consumption that purports to enhance cognitive function and also contains a botanical extract with established medicinal properties. The Federal Food, Drug, and Cosmetic Act (FDCA) defines a “drug” as an article intended to affect the structure or function of the body, and also articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease. A “food” is defined as an article used for food or drink for man or other animals, chewing gum, and articles used for components of any such article. The product in question is marketed as a dietary supplement, which is a category of food under the FDCA. However, the claims made about enhancing cognitive function and the presence of a botanical extract with known medicinal effects push it towards a drug classification. Specifically, if the product is intended to diagnose, cure, mitigate, treat, or prevent disease, or to affect the structure or function of the body in a manner beyond normal nutritional support, it would be considered a drug. The phrase “affect the structure or function of the body” is broad. While dietary supplements can make “structure/function claims” (e.g., “supports healthy joints”), these claims must be substantiated and cannot imply disease treatment or prevention. The presence of a botanical extract with documented medicinal properties, coupled with claims of cognitive enhancement, strongly suggests an intent to affect the body’s function in a way that likely crosses the line from a permissible dietary supplement claim to a drug claim. The regulatory burden for drugs is significantly higher, requiring premarket approval through a New Drug Application (NDA) or Biologics License Application (BLA). Dietary supplements, on the other hand, are regulated differently, with the manufacturer responsible for ensuring safety and proper labeling, and the FDA taking action against adulterated or misbranded products. Given the dual nature of the product’s claims and ingredients, the most appropriate regulatory pathway, and the one that aligns with the FDCA’s definitions, is to treat it as a drug. This necessitates an NDA process. The other options are incorrect because: classifying it solely as a dietary supplement ignores the drug-like claims and ingredients; treating it as a cosmetic is inappropriate as it is intended for ingestion and affects internal bodily functions, not external appearance; and the Public Health Service Act primarily governs biologics and certain public health activities, not the classification of food-like products with drug claims. Therefore, the product’s characteristics necessitate regulation as a drug, requiring an NDA.
-
Question 16 of 30
16. Question
A biotechnology firm has developed a novel, synthesized peptide designed to significantly extend the shelf-life of perishable baked goods by inhibiting microbial growth. This peptide is not naturally occurring in any foodstuff and has no documented history of safe consumption in food prior to its development. The firm intends to market this peptide as an ingredient that will be incorporated into baking formulations. Under the Federal Food, Drug, and Cosmetic Act (FDCA), what is the primary regulatory classification and required premarket action for this synthesized peptide?
Correct
The core of this question lies in understanding the regulatory distinctions between a food additive and a substance that is Generally Recognized as Safe (GRAS). A food additive, as defined by the Federal Food, Drug, and Cosmetic Act (FDCA), is any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food. This definition is broad and encompasses substances added for technological purposes during processing or storage. However, the FDCA also provides an exemption for substances that are GRAS for their intended use. GRAS status can be achieved through scientific procedures or, for substances used before 1958, through common use in food. The key differentiator is the regulatory pathway and the burden of proof. A substance intended for use as a food additive requires a food additive petition and FDA approval, demonstrating its safety under the conditions of its intended use. Conversely, a GRAS substance, whether through scientific procedures or common use, is exempt from the food additive petition process. The scenario describes a novel synthetic sweetener intended to enhance flavor. Without prior scientific procedures or common use history demonstrating its safety for this specific application, it would fall under the definition of a food additive. Therefore, the manufacturer would need to submit a food additive petition to the FDA for review and approval before marketing the sweetener. The explanation focuses on the statutory definitions and the procedural requirements mandated by the FDCA for substances intended for food use, highlighting the critical difference between a substance requiring premarket approval as an additive and one that is exempt due to established GRAS status. The correct approach is to recognize that a novel substance intended to affect food characteristics, without a pre-existing GRAS determination, necessitates a food additive petition.
Incorrect
The core of this question lies in understanding the regulatory distinctions between a food additive and a substance that is Generally Recognized as Safe (GRAS). A food additive, as defined by the Federal Food, Drug, and Cosmetic Act (FDCA), is any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food. This definition is broad and encompasses substances added for technological purposes during processing or storage. However, the FDCA also provides an exemption for substances that are GRAS for their intended use. GRAS status can be achieved through scientific procedures or, for substances used before 1958, through common use in food. The key differentiator is the regulatory pathway and the burden of proof. A substance intended for use as a food additive requires a food additive petition and FDA approval, demonstrating its safety under the conditions of its intended use. Conversely, a GRAS substance, whether through scientific procedures or common use, is exempt from the food additive petition process. The scenario describes a novel synthetic sweetener intended to enhance flavor. Without prior scientific procedures or common use history demonstrating its safety for this specific application, it would fall under the definition of a food additive. Therefore, the manufacturer would need to submit a food additive petition to the FDA for review and approval before marketing the sweetener. The explanation focuses on the statutory definitions and the procedural requirements mandated by the FDCA for substances intended for food use, highlighting the critical difference between a substance requiring premarket approval as an additive and one that is exempt due to established GRAS status. The correct approach is to recognize that a novel substance intended to affect food characteristics, without a pre-existing GRAS determination, necessitates a food additive petition.
-
Question 17 of 30
17. Question
A pharmaceutical company has developed “Neuro-Regen,” a novel compound showing promising preliminary results in Phase II clinical trials for a rare, debilitating neurological disorder for which no effective treatments currently exist. The company aims to accelerate the drug’s development and eventual market entry to address this significant unmet medical need. Considering the drug’s potential to offer substantial improvement over existing (albeit limited) supportive care, which FDA expedited program designation would be the most strategically advantageous for the company to pursue at this stage to facilitate intensive FDA guidance and a streamlined development process?
Correct
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for a rare neurological disorder. The manufacturer has completed Phase II clinical trials demonstrating preliminary efficacy and safety. To expedite market access for this critical unmet medical need, the manufacturer is considering regulatory pathways. The Food and Drug Administration (FDA) has several programs designed to accelerate the development and review of drugs for serious or life-threatening conditions. The options presented represent different FDA expedited programs. Breakthrough Therapy Designation is granted to drugs that demonstrate substantial improvement over available therapies based on preliminary clinical evidence. Fast Track designation is for drugs intended to treat serious conditions and that demonstrate the potential to address unmet medical needs. Accelerated Approval allows for earlier approval of drugs for serious conditions based on surrogate endpoints that are reasonably likely to predict clinical benefit, with post-approval confirmatory trials required. Priority Review is a designation that speeds up the FDA’s review of a drug application by shortening the target review time. Given that “Neuro-Regen” is for a rare neurological disorder (implying a serious condition) and has shown preliminary efficacy, the most appropriate initial consideration for expediting development and review, especially if it demonstrates substantial improvement over existing treatments, would be Breakthrough Therapy Designation. This designation provides intensive FDA guidance throughout the development process. While Fast Track is also relevant for serious conditions and unmet needs, Breakthrough Therapy is specifically for drugs showing substantial improvement. Accelerated Approval is typically considered when surrogate endpoints are used, which isn’t explicitly stated as the primary basis for early consideration here, and Priority Review is a review goal, not a development pathway designation. Therefore, Breakthrough Therapy Designation is the most fitting initial strategic regulatory consideration for a drug showing promising early results for a serious unmet medical need.
Incorrect
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for a rare neurological disorder. The manufacturer has completed Phase II clinical trials demonstrating preliminary efficacy and safety. To expedite market access for this critical unmet medical need, the manufacturer is considering regulatory pathways. The Food and Drug Administration (FDA) has several programs designed to accelerate the development and review of drugs for serious or life-threatening conditions. The options presented represent different FDA expedited programs. Breakthrough Therapy Designation is granted to drugs that demonstrate substantial improvement over available therapies based on preliminary clinical evidence. Fast Track designation is for drugs intended to treat serious conditions and that demonstrate the potential to address unmet medical needs. Accelerated Approval allows for earlier approval of drugs for serious conditions based on surrogate endpoints that are reasonably likely to predict clinical benefit, with post-approval confirmatory trials required. Priority Review is a designation that speeds up the FDA’s review of a drug application by shortening the target review time. Given that “Neuro-Regen” is for a rare neurological disorder (implying a serious condition) and has shown preliminary efficacy, the most appropriate initial consideration for expediting development and review, especially if it demonstrates substantial improvement over existing treatments, would be Breakthrough Therapy Designation. This designation provides intensive FDA guidance throughout the development process. While Fast Track is also relevant for serious conditions and unmet needs, Breakthrough Therapy is specifically for drugs showing substantial improvement. Accelerated Approval is typically considered when surrogate endpoints are used, which isn’t explicitly stated as the primary basis for early consideration here, and Priority Review is a review goal, not a development pathway designation. Therefore, Breakthrough Therapy Designation is the most fitting initial strategic regulatory consideration for a drug showing promising early results for a serious unmet medical need.
-
Question 18 of 30
18. Question
A pharmaceutical company has developed “Neuro-Regen,” a novel biologic intended to treat a rare, debilitating neurological disorder for which no effective treatments currently exist. Pre-clinical studies and early-stage human trials (Phase I and Phase II) have indicated a promising safety profile and a statistically significant improvement in a key biomarker that is reasonably likely to predict clinical benefit, although long-term clinical outcomes are still under investigation. The company wishes to make this therapy available to patients as soon as possible due to the severity of the condition. Which regulatory pathway, under the Federal Food, Drug, and Cosmetic Act (FDCA) and related guidance, would most appropriately facilitate the earliest possible market access for “Neuro-Regen” while acknowledging the need for post-market confirmation of clinical benefit?
Correct
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for a rare neurological disorder. The manufacturer has conducted Phase I and Phase II clinical trials demonstrating preliminary safety and efficacy. To expedite market access for this life-threatening condition, the manufacturer is considering pathways beyond the standard New Drug Application (NDA) process. The question asks about the most appropriate regulatory pathway for accelerated approval, considering the unmet medical need and the preliminary data. The Food and Drug Administration (FDA) offers several expedited programs to facilitate the development and review of drugs for serious or life-threatening conditions. These include Fast Track, Breakthrough Therapy, Accelerated Approval, and Priority Review. Fast Track designation allows for more frequent meetings with the FDA and eligibility for rolling review, but it does not guarantee approval. Breakthrough Therapy designation is for drugs that demonstrate substantial improvement over available therapy, requiring clear clinical evidence of superiority. Accelerated Approval allows for approval based on surrogate endpoints or intermediate clinical endpoints that are reasonably likely to predict clinical benefit, with post-marketing studies required to confirm the predicted benefit. Priority Review is a designation that shortens the FDA’s review goal from 10 months to 6 months for drugs that offer significant improvements in safety or efficacy. Given that the drug addresses a serious condition with an unmet medical need and has shown preliminary efficacy, but the full clinical benefit is not yet definitively established to warrant a standard approval or a breakthrough designation based on superiority, Accelerated Approval is the most fitting pathway. This pathway allows for earlier market entry based on surrogate endpoints, contingent on confirmatory trials. The other options are less suitable: Fast Track is a designation that facilitates development but doesn’t directly lead to earlier approval based on surrogate endpoints; Breakthrough Therapy requires a higher bar of demonstrated substantial improvement; and Priority Review is a review timeline designation, not a pathway for approval based on less definitive evidence.
Incorrect
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for a rare neurological disorder. The manufacturer has conducted Phase I and Phase II clinical trials demonstrating preliminary safety and efficacy. To expedite market access for this life-threatening condition, the manufacturer is considering pathways beyond the standard New Drug Application (NDA) process. The question asks about the most appropriate regulatory pathway for accelerated approval, considering the unmet medical need and the preliminary data. The Food and Drug Administration (FDA) offers several expedited programs to facilitate the development and review of drugs for serious or life-threatening conditions. These include Fast Track, Breakthrough Therapy, Accelerated Approval, and Priority Review. Fast Track designation allows for more frequent meetings with the FDA and eligibility for rolling review, but it does not guarantee approval. Breakthrough Therapy designation is for drugs that demonstrate substantial improvement over available therapy, requiring clear clinical evidence of superiority. Accelerated Approval allows for approval based on surrogate endpoints or intermediate clinical endpoints that are reasonably likely to predict clinical benefit, with post-marketing studies required to confirm the predicted benefit. Priority Review is a designation that shortens the FDA’s review goal from 10 months to 6 months for drugs that offer significant improvements in safety or efficacy. Given that the drug addresses a serious condition with an unmet medical need and has shown preliminary efficacy, but the full clinical benefit is not yet definitively established to warrant a standard approval or a breakthrough designation based on superiority, Accelerated Approval is the most fitting pathway. This pathway allows for earlier market entry based on surrogate endpoints, contingent on confirmatory trials. The other options are less suitable: Fast Track is a designation that facilitates development but doesn’t directly lead to earlier approval based on surrogate endpoints; Breakthrough Therapy requires a higher bar of demonstrated substantial improvement; and Priority Review is a review timeline designation, not a pathway for approval based on less definitive evidence.
-
Question 19 of 30
19. Question
A company manufactures a product called “Vita-Boost Elixir,” which is a complex blend of vitamins, minerals, and herbal extracts. The product is marketed with prominent claims on its packaging and website stating that it “enhances cellular regeneration” and “reverses age-related cellular damage.” The company asserts that the elixir is intended to support overall wellness and combat the effects of aging. Based on the Federal Food, Drug, and Cosmetic Act (FDCA), how would the Food and Drug Administration (FDA) most likely classify and regulate the “Vita-Boost Elixir” given these marketing claims?
Correct
The core of this question lies in understanding the regulatory distinction between a dietary supplement and a drug under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to treat, cure, mitigate, or prevent disease, or to affect the structure or function of the body in a way that goes beyond mere nutritional support, is considered a drug. The FDCA defines a drug as “articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals.” Furthermore, articles (other than food) intended to affect the structure or any function of the body of man or other animals are also considered drugs. In this scenario, the “Vita-Boost Elixir” is marketed with claims that it “enhances cellular regeneration” and “reverses age-related cellular damage.” These claims directly relate to affecting the structure and function of the body at a cellular level, and imply a therapeutic or preventative effect against aging, which is often considered a condition or disease state in a broad sense within regulatory frameworks. Therefore, the product, despite its formulation as a vitamin and mineral blend, falls under the definition of a drug. The Food and Drug Administration (FDA) would likely regulate it as such, requiring premarket approval through a New Drug Application (NDA) process, rather than through the less stringent regulations applicable to dietary supplements, which are governed by the Dietary Supplement Health and Education Act of 1994 (DSHEA). DSHEA defines dietary supplements as products taken by mouth that contain a dietary ingredient intended to supplement the diet and are intended to affect the structure or function of the body, but not to diagnose, treat, cure, or prevent any disease. The claims made for Vita-Boost Elixir exceed the permissible claims for dietary supplements by directly addressing cellular mechanisms and implying a restorative function that is characteristic of drug claims.
Incorrect
The core of this question lies in understanding the regulatory distinction between a dietary supplement and a drug under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to treat, cure, mitigate, or prevent disease, or to affect the structure or function of the body in a way that goes beyond mere nutritional support, is considered a drug. The FDCA defines a drug as “articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals.” Furthermore, articles (other than food) intended to affect the structure or any function of the body of man or other animals are also considered drugs. In this scenario, the “Vita-Boost Elixir” is marketed with claims that it “enhances cellular regeneration” and “reverses age-related cellular damage.” These claims directly relate to affecting the structure and function of the body at a cellular level, and imply a therapeutic or preventative effect against aging, which is often considered a condition or disease state in a broad sense within regulatory frameworks. Therefore, the product, despite its formulation as a vitamin and mineral blend, falls under the definition of a drug. The Food and Drug Administration (FDA) would likely regulate it as such, requiring premarket approval through a New Drug Application (NDA) process, rather than through the less stringent regulations applicable to dietary supplements, which are governed by the Dietary Supplement Health and Education Act of 1994 (DSHEA). DSHEA defines dietary supplements as products taken by mouth that contain a dietary ingredient intended to supplement the diet and are intended to affect the structure or function of the body, but not to diagnose, treat, cure, or prevent any disease. The claims made for Vita-Boost Elixir exceed the permissible claims for dietary supplements by directly addressing cellular mechanisms and implying a restorative function that is characteristic of drug claims.
-
Question 20 of 30
20. Question
A biotechnology firm develops a topical serum named “Revitalizing Elixir,” which is marketed with claims that it “enhances cellular regeneration” and “restores skin’s youthful elasticity by stimulating collagen production.” The product is packaged in a vial with a dropper for application directly to the skin. Based on the Federal Food, Drug, and Cosmetic Act (FDCA), how would the U.S. Food and Drug Administration (FDA) most likely classify this product and what would be the primary regulatory pathway for its market approval?
Correct
The core of this question lies in understanding the regulatory distinction between a “drug” and a “cosmetic” under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to affect the structure or function of the body, or to treat or prevent disease, is classified as a drug. Conversely, a product intended for cleansing, beautifying, promoting attractiveness, or altering the appearance without affecting the body’s structure or function is considered a cosmetic. In this scenario, the “Revitalizing Elixir” is marketed with claims that it “enhances cellular regeneration” and “restores skin’s youthful elasticity by stimulating collagen production.” These claims directly address the structure and function of the skin at a cellular level, indicating a therapeutic or physiological effect. Such claims push the product beyond mere beautification and into the realm of drug classification. Therefore, the elixir would be regulated as a drug, requiring an approved New Drug Application (NDA) before marketing, and subject to stringent Good Manufacturing Practices (GMPs) for drugs, as well as comprehensive labeling and advertising requirements applicable to pharmaceuticals. The other options represent incorrect classifications. Classifying it as a cosmetic would ignore the physiological claims. Labeling it as a dietary supplement would be inappropriate as it is applied topically and not ingested. Designating it as a medical device would also be incorrect, as it does not meet the definition of a device, which typically involves an instrument, apparatus, implement, machine, contrivance, implant, or other similar or related article, including any component, part, or accessory, which is intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals.
Incorrect
The core of this question lies in understanding the regulatory distinction between a “drug” and a “cosmetic” under the Federal Food, Drug, and Cosmetic Act (FDCA). A product intended to affect the structure or function of the body, or to treat or prevent disease, is classified as a drug. Conversely, a product intended for cleansing, beautifying, promoting attractiveness, or altering the appearance without affecting the body’s structure or function is considered a cosmetic. In this scenario, the “Revitalizing Elixir” is marketed with claims that it “enhances cellular regeneration” and “restores skin’s youthful elasticity by stimulating collagen production.” These claims directly address the structure and function of the skin at a cellular level, indicating a therapeutic or physiological effect. Such claims push the product beyond mere beautification and into the realm of drug classification. Therefore, the elixir would be regulated as a drug, requiring an approved New Drug Application (NDA) before marketing, and subject to stringent Good Manufacturing Practices (GMPs) for drugs, as well as comprehensive labeling and advertising requirements applicable to pharmaceuticals. The other options represent incorrect classifications. Classifying it as a cosmetic would ignore the physiological claims. Labeling it as a dietary supplement would be inappropriate as it is applied topically and not ingested. Designating it as a medical device would also be incorrect, as it does not meet the definition of a device, which typically involves an instrument, apparatus, implement, machine, contrivance, implant, or other similar or related article, including any component, part, or accessory, which is intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals.
-
Question 21 of 30
21. Question
A pharmaceutical company has developed “Neuro-Regen,” a novel biologic intended to treat a rare, debilitating neurodegenerative disease for which no effective treatments currently exist. Pre-clinical studies and early-stage human trials (Phase I and II) have demonstrated promising results, indicating a potential for significant disease modification and symptom alleviation. The preliminary data suggests that Neuro-Regen may substantially improve upon the limited symptomatic relief offered by existing palliative care. The company is eager to make this therapy available to patients as quickly as possible, recognizing the severe unmet medical need. Which regulatory pathway, under the Federal Food, Drug, and Cosmetic Act (FDCA) and its amendments, would most appropriately facilitate earlier market entry for Neuro-Regen, contingent upon the submission of robust post-market confirmatory studies to verify the anticipated clinical benefit?
Correct
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for treating a rare neurodegenerative disorder. The developer has completed Phase II clinical trials demonstrating preliminary efficacy and safety. To expedite market access for this unmet medical need, the developer seeks a regulatory pathway that balances timely availability with robust post-market oversight. The Food and Drug Administration (FDA) has several expedited programs, including Fast Track, Breakthrough Therapy, Accelerated Approval, and Priority Review. Fast Track designation facilitates more frequent communication between the FDA and the sponsor and allows for rolling review of the marketing application. Breakthrough Therapy designation is for drugs that treat a serious or life-threatening condition and preliminary clinical evidence indicates that the drug may be substantially better than available therapies. Accelerated Approval allows for approval based on surrogate or intermediate clinical endpoints that are reasonably likely to predict clinical benefit, with a requirement for post-approval confirmatory trials. Priority Review is a designation that shortens the FDA’s review timeline for certain drugs that offer significant improvements in safety or efficacy. Given that Neuro-Regen addresses a rare, serious condition and preliminary data suggests potential for substantial improvement over existing treatments (even if not definitively proven superior yet), and the goal is to expedite availability, the most appropriate pathway that combines early access with a clear mandate for further evidence generation is Accelerated Approval. This pathway is specifically designed for situations where a drug may fill an unmet need and can be approved based on surrogate endpoints, provided confirmatory trials are committed. While Breakthrough Therapy could be sought, Accelerated Approval directly addresses the mechanism of getting the drug to patients sooner based on a strong likelihood of benefit, contingent on post-market validation. Fast Track and Priority Review are more about the process of review and communication, not the basis of approval itself in the context of potentially limited definitive data. The question asks for the pathway that allows for earlier market entry based on a reasonable likelihood of clinical benefit, which is the hallmark of Accelerated Approval.
Incorrect
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended for treating a rare neurodegenerative disorder. The developer has completed Phase II clinical trials demonstrating preliminary efficacy and safety. To expedite market access for this unmet medical need, the developer seeks a regulatory pathway that balances timely availability with robust post-market oversight. The Food and Drug Administration (FDA) has several expedited programs, including Fast Track, Breakthrough Therapy, Accelerated Approval, and Priority Review. Fast Track designation facilitates more frequent communication between the FDA and the sponsor and allows for rolling review of the marketing application. Breakthrough Therapy designation is for drugs that treat a serious or life-threatening condition and preliminary clinical evidence indicates that the drug may be substantially better than available therapies. Accelerated Approval allows for approval based on surrogate or intermediate clinical endpoints that are reasonably likely to predict clinical benefit, with a requirement for post-approval confirmatory trials. Priority Review is a designation that shortens the FDA’s review timeline for certain drugs that offer significant improvements in safety or efficacy. Given that Neuro-Regen addresses a rare, serious condition and preliminary data suggests potential for substantial improvement over existing treatments (even if not definitively proven superior yet), and the goal is to expedite availability, the most appropriate pathway that combines early access with a clear mandate for further evidence generation is Accelerated Approval. This pathway is specifically designed for situations where a drug may fill an unmet need and can be approved based on surrogate endpoints, provided confirmatory trials are committed. While Breakthrough Therapy could be sought, Accelerated Approval directly addresses the mechanism of getting the drug to patients sooner based on a strong likelihood of benefit, contingent on post-market validation. Fast Track and Priority Review are more about the process of review and communication, not the basis of approval itself in the context of potentially limited definitive data. The question asks for the pathway that allows for earlier market entry based on a reasonable likelihood of clinical benefit, which is the hallmark of Accelerated Approval.
-
Question 22 of 30
22. Question
A biotechnology firm has successfully engineered a novel therapeutic agent that involves the direct administration of modified human cells designed to target and destroy specific cancer cells. This agent is not a synthesized chemical compound, nor is it a vaccine or a blood derivative. It represents a significant advancement in cellular therapy. Considering the regulatory framework for biological products and drugs in the United States, which regulatory pathway would be the primary mechanism for seeking marketing approval for this innovative treatment?
Correct
The scenario describes a situation where a novel therapeutic agent, developed through advanced genetic engineering, is intended for human use. This agent is not a traditional small molecule drug or a purified protein, but rather a complex biological product that leverages cellular mechanisms for its therapeutic effect. The critical regulatory pathway for such a product, especially one that is not a vaccine or blood product but rather a cell-based therapy or a gene therapy, falls under the purview of the Biologics License Application (BLA) process. The BLA is specifically designed for biological products, which are generally derived from living organisms and are often more complex and variable than chemically synthesized drugs. While the Investigational New Drug (IND) application is a prerequisite for clinical testing, the ultimate approval for marketing a biologic rests on the BLA. The New Drug Application (NDA) is reserved for conventional drugs. Premarket Notification (510(k)) and Premarket Approval (PMA) are pathways for medical devices, not biological products. Therefore, the most appropriate regulatory pathway for this genetically engineered therapeutic agent, given its nature as a biological product, is the BLA.
Incorrect
The scenario describes a situation where a novel therapeutic agent, developed through advanced genetic engineering, is intended for human use. This agent is not a traditional small molecule drug or a purified protein, but rather a complex biological product that leverages cellular mechanisms for its therapeutic effect. The critical regulatory pathway for such a product, especially one that is not a vaccine or blood product but rather a cell-based therapy or a gene therapy, falls under the purview of the Biologics License Application (BLA) process. The BLA is specifically designed for biological products, which are generally derived from living organisms and are often more complex and variable than chemically synthesized drugs. While the Investigational New Drug (IND) application is a prerequisite for clinical testing, the ultimate approval for marketing a biologic rests on the BLA. The New Drug Application (NDA) is reserved for conventional drugs. Premarket Notification (510(k)) and Premarket Approval (PMA) are pathways for medical devices, not biological products. Therefore, the most appropriate regulatory pathway for this genetically engineered therapeutic agent, given its nature as a biological product, is the BLA.
-
Question 23 of 30
23. Question
A company develops a novel beverage, “Vita-Boost Elixir,” composed of a proprietary blend of vitamins, minerals, and botanical extracts. The product is marketed with prominent claims on its packaging and website stating it “significantly enhances cognitive function in adults” and “effectively reduces stress-related fatigue.” While the ingredients are commonly found in dietary supplements and foods, the company’s marketing strategy explicitly targets individuals seeking to improve mental acuity and manage the physiological effects of stress. Under the Federal Food, Drug, and Cosmetic Act (FDCA), how would the U.S. Food and Drug Administration (FDA) most likely classify and regulate the “Vita-Boost Elixir” given these marketing claims and its composition?
Correct
The core issue revolves around the regulatory classification of a product that exhibits characteristics of both a food and a drug. The Federal Food, Drug, and Cosmetic Act (FDCA) defines “drug” as articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in humans or other animals, and articles (other than food) intended to affect the structure or any function of the body. A “food” is defined as an article used for food or drink for man or other animals, chewing gum, and articles used for components of any such article. When a product is marketed with claims that bring it within the definition of a drug, it is regulated as a drug, even if it also meets the definition of a food. In this scenario, the “Vita-Boost Elixir” is marketed with claims of “enhancing cognitive function” and “reducing stress-related fatigue,” which directly relate to affecting the structure or function of the body and mitigating disease symptoms (stress-related fatigue can be considered a symptom of a condition). Therefore, despite its composition as a blend of vitamins and herbal extracts, the intended use, as evidenced by its labeling claims, places it squarely within the regulatory purview of the FDA as a drug. The Food Safety Modernization Act (FSMA) primarily focuses on preventive controls for foodborne illness and does not alter the fundamental classification of a product intended to treat or prevent disease. While the product might also need to comply with food labeling requirements if it were solely marketed as a food, the drug claims trigger the more stringent drug regulatory pathway, including the requirement for an approved New Drug Application (NDA) or Investigational New Drug (IND) application for clinical testing. The question tests the understanding of how intended use and claims dictate regulatory classification under the FDCA, particularly the distinction between food and drug. The correct classification hinges on the product’s marketing and claims, not solely its ingredients.
Incorrect
The core issue revolves around the regulatory classification of a product that exhibits characteristics of both a food and a drug. The Federal Food, Drug, and Cosmetic Act (FDCA) defines “drug” as articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in humans or other animals, and articles (other than food) intended to affect the structure or any function of the body. A “food” is defined as an article used for food or drink for man or other animals, chewing gum, and articles used for components of any such article. When a product is marketed with claims that bring it within the definition of a drug, it is regulated as a drug, even if it also meets the definition of a food. In this scenario, the “Vita-Boost Elixir” is marketed with claims of “enhancing cognitive function” and “reducing stress-related fatigue,” which directly relate to affecting the structure or function of the body and mitigating disease symptoms (stress-related fatigue can be considered a symptom of a condition). Therefore, despite its composition as a blend of vitamins and herbal extracts, the intended use, as evidenced by its labeling claims, places it squarely within the regulatory purview of the FDA as a drug. The Food Safety Modernization Act (FSMA) primarily focuses on preventive controls for foodborne illness and does not alter the fundamental classification of a product intended to treat or prevent disease. While the product might also need to comply with food labeling requirements if it were solely marketed as a food, the drug claims trigger the more stringent drug regulatory pathway, including the requirement for an approved New Drug Application (NDA) or Investigational New Drug (IND) application for clinical testing. The question tests the understanding of how intended use and claims dictate regulatory classification under the FDCA, particularly the distinction between food and drug. The correct classification hinges on the product’s marketing and claims, not solely its ingredients.
-
Question 24 of 30
24. Question
A biotechnology firm develops a novel oral capsule containing a proprietary blend of amino acids and botanical extracts. The product is marketed with claims that it “enhances mental acuity by modulating neurotransmitter receptor binding in the prefrontal cortex.” The company asserts it is a dietary supplement. Based on the Federal Food, Drug, and Cosmetic Act (FDCA) and relevant regulatory interpretations, what is the most accurate classification and required regulatory pathway for this product if it were to be marketed in the United States?
Correct
The core issue revolves around the regulatory classification of a product intended for oral consumption that claims to enhance cognitive function by altering brain chemistry, specifically by modulating neurotransmitter receptor binding. Under the Federal Food, Drug, and Cosmetic Act (FDCA), a product intended to affect the structure or function of the body, or intended to diagnose, cure, mitigate, treat, or prevent disease, is considered a drug. The claim of “modulating neurotransmitter receptor binding” directly implicates the structure and function of the body, specifically the nervous system. While the product is presented as a dietary supplement, its intended use and claims push it beyond the scope of dietary supplements, which are intended to supplement the diet and bear structure/function claims that do not relate to disease or specific bodily functions in a drug-like manner. The Food and Drug Administration (FDA) would likely view such claims and intended use as making the product an unapproved new drug. Therefore, the most appropriate regulatory pathway and classification would be as a drug, requiring an approved New Drug Application (NDA) before marketing. The other options are less fitting: a dietary supplement typically does not make claims about altering specific receptor binding; a cosmetic is intended for application to the external parts of the body or the teeth and mouth, and does not affect the structure or function of the body; and a medical device is an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals. This product, being orally ingested and acting on internal biological processes, does not fit the definition of a medical device.
Incorrect
The core issue revolves around the regulatory classification of a product intended for oral consumption that claims to enhance cognitive function by altering brain chemistry, specifically by modulating neurotransmitter receptor binding. Under the Federal Food, Drug, and Cosmetic Act (FDCA), a product intended to affect the structure or function of the body, or intended to diagnose, cure, mitigate, treat, or prevent disease, is considered a drug. The claim of “modulating neurotransmitter receptor binding” directly implicates the structure and function of the body, specifically the nervous system. While the product is presented as a dietary supplement, its intended use and claims push it beyond the scope of dietary supplements, which are intended to supplement the diet and bear structure/function claims that do not relate to disease or specific bodily functions in a drug-like manner. The Food and Drug Administration (FDA) would likely view such claims and intended use as making the product an unapproved new drug. Therefore, the most appropriate regulatory pathway and classification would be as a drug, requiring an approved New Drug Application (NDA) before marketing. The other options are less fitting: a dietary supplement typically does not make claims about altering specific receptor binding; a cosmetic is intended for application to the external parts of the body or the teeth and mouth, and does not affect the structure or function of the body; and a medical device is an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals. This product, being orally ingested and acting on internal biological processes, does not fit the definition of a medical device.
-
Question 25 of 30
25. Question
A biotechnology firm develops an ingestible product containing a genetically modified strain of yeast engineered to produce a specific B vitamin precursor directly within the human digestive tract. The company intends to market this product as a dietary supplement to enhance nutrient absorption. Considering the regulatory definitions under the Federal Food, Drug, and Cosmetic Act (FDCA) and the Food Safety Modernization Act (FSMA), what is the most appropriate initial regulatory pathway for this product?
Correct
The core issue revolves around the regulatory classification of a novel ingestible product containing genetically modified yeast designed to produce a specific vitamin precursor. The manufacturer claims it functions as a dietary supplement. However, the presence of genetically modified organisms (GMOs) and the intended mechanism of action (in-vivo synthesis of a precursor) necessitate a careful examination of the Federal Food, Drug, and Cosmetic Act (FDCA) and related regulations. Under the FDCA, a “drug” is defined as an article intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals, or articles (other than food) intended to affect the structure or any function of the body of man or other animals. A “food” is defined as an article used for food or drink for man or other animals, chewing gum, and articles used for components of any such article. Dietary supplements are a category of food, regulated under specific provisions of the FDCA, including those related to labeling and good manufacturing practices. The critical distinction here lies in the product’s intended use and its mechanism of action. While the manufacturer markets it as a dietary supplement (food), the genetically modified yeast’s engineered function to synthesize a vitamin precursor within the body goes beyond the typical definition of a food ingredient. This engineered biological activity, aimed at influencing bodily function (vitamin synthesis), strongly suggests it could be regulated as a drug if it meets the criteria for affecting the structure or function of the body in a manner intended for therapeutic or physiological modification. The presence of GMOs, while not automatically classifying it as a drug, adds complexity and may trigger additional regulatory scrutiny under various provisions, including those related to food safety and potentially new dietary ingredient notifications if it were to remain solely within the food classification. However, the engineered biological function is the primary driver for considering a drug classification. The question asks for the *most appropriate* regulatory pathway. Given the engineered biological function to synthesize a precursor, which directly affects a bodily function (vitamin production), the product likely falls under the definition of a drug, requiring an Investigational New Drug (IND) application for clinical testing and a New Drug Application (NDA) for marketing approval, rather than simply a dietary supplement notification or a food additive petition. The Food Safety Modernization Act (FSMA) primarily addresses food safety and preventive controls for foodborne illnesses, and while relevant for any food product, it doesn’t supersede the drug classification if the product’s intended use and mechanism align with the drug definition. The Public Health Service Act (PHSA) primarily governs biologics, vaccines, and blood products, which this product, as a genetically modified yeast, might also be considered, but the initial pathway for a novel ingestible product with a specific physiological effect is often assessed through the drug pathway first. Therefore, the most encompassing and appropriate initial regulatory pathway for a product with an engineered biological function affecting the body’s processes, even if marketed as a supplement, is the drug pathway.
Incorrect
The core issue revolves around the regulatory classification of a novel ingestible product containing genetically modified yeast designed to produce a specific vitamin precursor. The manufacturer claims it functions as a dietary supplement. However, the presence of genetically modified organisms (GMOs) and the intended mechanism of action (in-vivo synthesis of a precursor) necessitate a careful examination of the Federal Food, Drug, and Cosmetic Act (FDCA) and related regulations. Under the FDCA, a “drug” is defined as an article intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals, or articles (other than food) intended to affect the structure or any function of the body of man or other animals. A “food” is defined as an article used for food or drink for man or other animals, chewing gum, and articles used for components of any such article. Dietary supplements are a category of food, regulated under specific provisions of the FDCA, including those related to labeling and good manufacturing practices. The critical distinction here lies in the product’s intended use and its mechanism of action. While the manufacturer markets it as a dietary supplement (food), the genetically modified yeast’s engineered function to synthesize a vitamin precursor within the body goes beyond the typical definition of a food ingredient. This engineered biological activity, aimed at influencing bodily function (vitamin synthesis), strongly suggests it could be regulated as a drug if it meets the criteria for affecting the structure or function of the body in a manner intended for therapeutic or physiological modification. The presence of GMOs, while not automatically classifying it as a drug, adds complexity and may trigger additional regulatory scrutiny under various provisions, including those related to food safety and potentially new dietary ingredient notifications if it were to remain solely within the food classification. However, the engineered biological function is the primary driver for considering a drug classification. The question asks for the *most appropriate* regulatory pathway. Given the engineered biological function to synthesize a precursor, which directly affects a bodily function (vitamin production), the product likely falls under the definition of a drug, requiring an Investigational New Drug (IND) application for clinical testing and a New Drug Application (NDA) for marketing approval, rather than simply a dietary supplement notification or a food additive petition. The Food Safety Modernization Act (FSMA) primarily addresses food safety and preventive controls for foodborne illnesses, and while relevant for any food product, it doesn’t supersede the drug classification if the product’s intended use and mechanism align with the drug definition. The Public Health Service Act (PHSA) primarily governs biologics, vaccines, and blood products, which this product, as a genetically modified yeast, might also be considered, but the initial pathway for a novel ingestible product with a specific physiological effect is often assessed through the drug pathway first. Therefore, the most encompassing and appropriate initial regulatory pathway for a product with an engineered biological function affecting the body’s processes, even if marketed as a supplement, is the drug pathway.
-
Question 26 of 30
26. Question
A company develops a novel botanical extract purported to lower blood pressure and also contains essential vitamins. The product is marketed with prominent claims on its packaging stating, “Helps manage hypertension and supports cardiovascular health,” alongside a list of its vitamin content. The company intends to sell this product directly to consumers without seeking premarket approval from the Food and Drug Administration (FDA). Under the Federal Food, Drug, and Cosmetic Act (FDCA), how would the FDA most likely classify this product, and what would be the primary regulatory implication?
Correct
The core issue revolves around the regulatory classification of a product intended for both nutritional supplementation and the treatment of a specific medical condition. The Federal Food, Drug, and Cosmetic Act (FDCA) distinguishes between food, drugs, and dietary supplements. A product that makes claims to diagnose, cure, mitigate, treat, or prevent disease falls under the definition of a drug, regardless of its nutritional content or intended use as a supplement. The manufacturer’s explicit claims on the product labeling and marketing materials are paramount in this determination. If the product is marketed with claims to alleviate symptoms of a diagnosed medical condition, such as hypertension, it is considered a drug and must undergo the rigorous premarket approval process, including an approved New Drug Application (NDA), before it can be legally marketed. Failure to do so renders the product adulterated and misbranded under the FDCA. Therefore, the product’s intended use, as evidenced by its claims, dictates its regulatory pathway. The fact that it also contains vitamins and minerals does not exempt it from drug regulations if it also purports to treat a disease.
Incorrect
The core issue revolves around the regulatory classification of a product intended for both nutritional supplementation and the treatment of a specific medical condition. The Federal Food, Drug, and Cosmetic Act (FDCA) distinguishes between food, drugs, and dietary supplements. A product that makes claims to diagnose, cure, mitigate, treat, or prevent disease falls under the definition of a drug, regardless of its nutritional content or intended use as a supplement. The manufacturer’s explicit claims on the product labeling and marketing materials are paramount in this determination. If the product is marketed with claims to alleviate symptoms of a diagnosed medical condition, such as hypertension, it is considered a drug and must undergo the rigorous premarket approval process, including an approved New Drug Application (NDA), before it can be legally marketed. Failure to do so renders the product adulterated and misbranded under the FDCA. Therefore, the product’s intended use, as evidenced by its claims, dictates its regulatory pathway. The fact that it also contains vitamins and minerals does not exempt it from drug regulations if it also purports to treat a disease.
-
Question 27 of 30
27. Question
A biotechnology firm, “AromaGen Innovations,” has developed a novel synthetic compound intended to impart a unique savory umami flavor to plant-based meat alternatives. This compound has no documented history of consumption in food prior to January 1, 1958, and the company has not yet conducted the comprehensive scientific studies required to establish its safety through scientific procedures. AromaGen Innovations intends to market this compound as a direct ingredient in their client’s food products. Under the Federal Food, Drug, and Cosmetic Act (FDCA), what regulatory pathway is most appropriate for AromaGen Innovations to ensure the legal marketing of this new flavoring agent?
Correct
The core of this question lies in understanding the regulatory distinction between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance that is intended for use in food and does not meet the criteria for GRAS status, either through scientific procedures or through experience based on common use in food prior to January 1, 1958, is considered a food additive. As such, it requires premarket approval through a Food Additive Petition (FAP) process, culminating in the issuance of a regulation by the FDA. This process ensures that the substance is safe for its intended use under the conditions of use specified in the regulation. Conversely, if a substance has a history of common use in food prior to 1958 or has been affirmed as GRAS through scientific procedures, it does not require premarket approval as a food additive. The scenario describes a novel flavoring agent with no prior history of food use, and its safety has not been established through scientific procedures. Therefore, it must undergo the rigorous premarket review and approval process applicable to food additives. The absence of a prior FAP or a GRAS affirmation means it falls squarely into the category requiring a new petition. The correct approach is to recognize that the lack of established safety data and prior use necessitates a formal regulatory pathway for approval, which is the Food Additive Petition.
Incorrect
The core of this question lies in understanding the regulatory distinction between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance that is intended for use in food and does not meet the criteria for GRAS status, either through scientific procedures or through experience based on common use in food prior to January 1, 1958, is considered a food additive. As such, it requires premarket approval through a Food Additive Petition (FAP) process, culminating in the issuance of a regulation by the FDA. This process ensures that the substance is safe for its intended use under the conditions of use specified in the regulation. Conversely, if a substance has a history of common use in food prior to 1958 or has been affirmed as GRAS through scientific procedures, it does not require premarket approval as a food additive. The scenario describes a novel flavoring agent with no prior history of food use, and its safety has not been established through scientific procedures. Therefore, it must undergo the rigorous premarket review and approval process applicable to food additives. The absence of a prior FAP or a GRAS affirmation means it falls squarely into the category requiring a new petition. The correct approach is to recognize that the lack of established safety data and prior use necessitates a formal regulatory pathway for approval, which is the Food Additive Petition.
-
Question 28 of 30
28. Question
A company introduces a new skincare product, “Radiant Glow Serum,” which is advertised with claims that it “visibly reduces the appearance of fine lines and wrinkles by stimulating natural collagen production and significantly improving skin elasticity.” The product’s active ingredient is a proprietary peptide complex. Considering the regulatory definitions under the Federal Food, Drug, and Cosmetic Act, how would the U.S. Food and Drug Administration (FDA) most likely classify this product based on its marketing claims?
Correct
The core of this question lies in understanding the regulatory distinction between a drug and a cosmetic under the Federal Food, Drug, and Cosmetic Act (FD&C Act). A product intended to affect the structure or function of the body, or to diagnose, cure, mitigate, treat, or prevent disease, is classified as a drug. Conversely, a cosmetic is intended for cleansing, beautifying, promoting attractiveness, or altering the appearance without affecting the body’s structure or function. In this scenario, the “Radiant Glow Serum” is marketed with claims of reducing the appearance of fine lines and wrinkles by stimulating collagen production and increasing skin elasticity. These claims directly relate to altering the structure and function of the skin, specifically its aging process and physical properties. Therefore, the serum would be regulated as a drug. The FD&C Act, specifically Section 201(g)(1)(B), defines a drug as an article intended to affect the structure or any function of the body of man or other animals. The claims made for the serum fall squarely within this definition. The regulatory pathway for such a product would necessitate an approved New Drug Application (NDA) or an Investigational New Drug (IND) application if it were a new molecular entity undergoing clinical trials. The absence of such approval, coupled with these drug-like claims, would render the product misbranded and adulterated, subject to FDA enforcement actions.
Incorrect
The core of this question lies in understanding the regulatory distinction between a drug and a cosmetic under the Federal Food, Drug, and Cosmetic Act (FD&C Act). A product intended to affect the structure or function of the body, or to diagnose, cure, mitigate, treat, or prevent disease, is classified as a drug. Conversely, a cosmetic is intended for cleansing, beautifying, promoting attractiveness, or altering the appearance without affecting the body’s structure or function. In this scenario, the “Radiant Glow Serum” is marketed with claims of reducing the appearance of fine lines and wrinkles by stimulating collagen production and increasing skin elasticity. These claims directly relate to altering the structure and function of the skin, specifically its aging process and physical properties. Therefore, the serum would be regulated as a drug. The FD&C Act, specifically Section 201(g)(1)(B), defines a drug as an article intended to affect the structure or any function of the body of man or other animals. The claims made for the serum fall squarely within this definition. The regulatory pathway for such a product would necessitate an approved New Drug Application (NDA) or an Investigational New Drug (IND) application if it were a new molecular entity undergoing clinical trials. The absence of such approval, coupled with these drug-like claims, would render the product misbranded and adulterated, subject to FDA enforcement actions.
-
Question 29 of 30
29. Question
Consider a novel, synthesized compound, “Chrono-Stabilizer X,” developed by a food manufacturer to significantly extend the shelf-life of baked goods by preventing lipid oxidation. This compound is intentionally incorporated into the dough formulation at a concentration of 0.05% by weight. Extensive internal laboratory studies have demonstrated its efficacy and have provided preliminary data suggesting no adverse toxicological effects at anticipated consumption levels. However, this compound has no history of common use in food prior to 1958, and the manufacturer has not submitted a GRAS notification to the FDA, nor is there a broad scientific consensus regarding its safety for human consumption. Under the Federal Food, Drug, and Cosmetic Act, what regulatory pathway is most appropriate for Chrono-Stabilizer X to be legally used in food products?
Correct
The core of this question lies in understanding the regulatory distinctions between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance that is intentionally added to food and performs a technological function is generally considered a food additive. For such substances, premarket approval is typically required, involving a rigorous scientific review to ensure safety under intended conditions of use. The FDCA defines a food additive as “any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food.” This definition is broad and encompasses substances added for preservation, texture, color, flavor, or nutritional enhancement. In contrast, a substance that is GRAS is exempt from the definition of a food additive. This exemption is based on a history of common use in food prior to 1958 or on scientific procedures that establish its safety. The determination of GRAS status can be made by the FDA through a formal notification process or by the food industry itself, provided there is a broad consensus of scientific opinion on its safety. The key differentiator is the requirement for premarket approval. Food additives require it; GRAS substances do not, although the FDA retains the authority to review GRAS determinations. Therefore, a substance that is intentionally added to food for a specific technological purpose, and for which no prior GRAS determination or scientific consensus on safety exists, would fall under the food additive regulations, necessitating a Food Additive Petition (FAP).
Incorrect
The core of this question lies in understanding the regulatory distinctions between a food additive and a GRAS (Generally Recognized as Safe) substance under the Federal Food, Drug, and Cosmetic Act (FDCA). A substance that is intentionally added to food and performs a technological function is generally considered a food additive. For such substances, premarket approval is typically required, involving a rigorous scientific review to ensure safety under intended conditions of use. The FDCA defines a food additive as “any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food.” This definition is broad and encompasses substances added for preservation, texture, color, flavor, or nutritional enhancement. In contrast, a substance that is GRAS is exempt from the definition of a food additive. This exemption is based on a history of common use in food prior to 1958 or on scientific procedures that establish its safety. The determination of GRAS status can be made by the FDA through a formal notification process or by the food industry itself, provided there is a broad consensus of scientific opinion on its safety. The key differentiator is the requirement for premarket approval. Food additives require it; GRAS substances do not, although the FDA retains the authority to review GRAS determinations. Therefore, a substance that is intentionally added to food for a specific technological purpose, and for which no prior GRAS determination or scientific consensus on safety exists, would fall under the food additive regulations, necessitating a Food Additive Petition (FAP).
-
Question 30 of 30
30. Question
A pharmaceutical company has developed “Neuro-Regen,” a promising new drug for a rare neurodegenerative condition affecting fewer than 5,000 individuals in the U.S. Pre-clinical studies and early-stage human trials (Phase I and Phase II) have demonstrated a positive safety profile and initial signs of therapeutic efficacy. The company aims to expedite the availability of this treatment to patients with a significant unmet medical need. Which combination of regulatory designations and pathways would most strategically align with the goal of efficient market access for “Neuro-Regen” under the Federal Food, Drug, and Cosmetic Act?
Correct
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended to treat a rare neurodegenerative disorder. The manufacturer has conducted extensive pre-clinical studies demonstrating safety and potential efficacy, followed by Phase I and Phase II clinical trials that established a favorable safety profile and preliminary evidence of therapeutic benefit in a small patient cohort. The disorder affects fewer than 5,000 individuals in the United States. The manufacturer is seeking the most efficient regulatory pathway to bring this treatment to market, acknowledging the significant unmet medical need. The Food, Drug, and Cosmetic Act (FDCA), as amended, provides several mechanisms for expedited review of drugs that address serious or life-threatening conditions. Specifically, the Orphan Drug Act of 1983 grants incentives for the development of drugs for rare diseases. Furthermore, the FDA has established programs like Fast Track, Breakthrough Therapy, Accelerated Approval, and Priority Review to expedite the development and review of drugs for serious conditions. Considering the rarity of the disease and the preliminary evidence of efficacy, the most appropriate regulatory strategy would involve leveraging the Orphan Drug designation, which provides market exclusivity and other incentives. Simultaneously, pursuing a Breakthrough Therapy designation would allow for more intensive FDA guidance and a potentially expedited review process, including the possibility of utilizing data from earlier phases of clinical development if they meet the criteria for accelerated approval. While Fast Track designation also offers expedited review, Breakthrough Therapy designation is generally considered more advantageous for drugs with strong early clinical evidence of substantial improvement over existing therapies. Accelerated Approval allows for earlier market entry based on surrogate endpoints, with post-marketing studies required to confirm clinical benefit. Priority Review is a designation that shortens the review timeline for drugs that offer significant improvements. Given the information, a combination of Orphan Drug designation for market exclusivity and incentives, coupled with a Breakthrough Therapy designation to facilitate development and review, and potentially seeking Accelerated Approval based on robust Phase II data if surrogate endpoints are well-established and clinically meaningful for this rare condition, represents the most comprehensive and advantageous regulatory approach. This strategy maximizes the chances of early market access while adhering to the rigorous scientific standards of the FDA.
Incorrect
The scenario describes a novel therapeutic agent, “Neuro-Regen,” intended to treat a rare neurodegenerative disorder. The manufacturer has conducted extensive pre-clinical studies demonstrating safety and potential efficacy, followed by Phase I and Phase II clinical trials that established a favorable safety profile and preliminary evidence of therapeutic benefit in a small patient cohort. The disorder affects fewer than 5,000 individuals in the United States. The manufacturer is seeking the most efficient regulatory pathway to bring this treatment to market, acknowledging the significant unmet medical need. The Food, Drug, and Cosmetic Act (FDCA), as amended, provides several mechanisms for expedited review of drugs that address serious or life-threatening conditions. Specifically, the Orphan Drug Act of 1983 grants incentives for the development of drugs for rare diseases. Furthermore, the FDA has established programs like Fast Track, Breakthrough Therapy, Accelerated Approval, and Priority Review to expedite the development and review of drugs for serious conditions. Considering the rarity of the disease and the preliminary evidence of efficacy, the most appropriate regulatory strategy would involve leveraging the Orphan Drug designation, which provides market exclusivity and other incentives. Simultaneously, pursuing a Breakthrough Therapy designation would allow for more intensive FDA guidance and a potentially expedited review process, including the possibility of utilizing data from earlier phases of clinical development if they meet the criteria for accelerated approval. While Fast Track designation also offers expedited review, Breakthrough Therapy designation is generally considered more advantageous for drugs with strong early clinical evidence of substantial improvement over existing therapies. Accelerated Approval allows for earlier market entry based on surrogate endpoints, with post-marketing studies required to confirm clinical benefit. Priority Review is a designation that shortens the review timeline for drugs that offer significant improvements. Given the information, a combination of Orphan Drug designation for market exclusivity and incentives, coupled with a Breakthrough Therapy designation to facilitate development and review, and potentially seeking Accelerated Approval based on robust Phase II data if surrogate endpoints are well-established and clinically meaningful for this rare condition, represents the most comprehensive and advantageous regulatory approach. This strategy maximizes the chances of early market access while adhering to the rigorous scientific standards of the FDA.